 2015, Vol. 35
2015, Vol. 35文章信息
- 丁一, 吴海英, 史吉平, 孙俊松
- DING Yi, WU Hai-ying, SHI Ji-ping, SUN Jun-song
- 氢化酶重组表达研究进展
- Current Progress in Recombinant Systems for Expression of Hydrogenases
- 中国生物工程杂志, 2015, 35(5): 109-118
- China Biotechnology, 2015, 35(5): 109-118
- http://dx.doi.org/10.13523/j.cb.20150516
-
文章历史
- 收稿日期:2015-02-03
- 修回日期:2015-03-14






2. 上海科技大学生命科学学院 上海 201210;
3. 中国科学院大学 北京 100049
2. School of Life Science and Technology, Shanghai Tech University, Shanghai 201210, China;
3. University of Chinese Academy of Sciences, Beijing 100049, China
20世纪以来,随着全球经济的迅猛发展,世界各国对能源的需求量日趋增大,而作为主要能源的化石燃料的使用带来了严重的环境问题,使得清洁可再生能源的开发迫在眉睫。氢气作为一种理想的可再生能源,受到广泛重视。与传统能源相比,氢气具有很多优点:它的燃烧热值高,达到142 KJ/g,是汽油的三倍(汽油47.3 KJ/g,甲烷55.5 KJ/g);其燃烧排放仅为水和少量氮氢化合物,是最清洁的能源;氢气燃烧及排放无腐蚀性,对设备无损。但目前工业制氢方法却主要是矿物燃料制氢,包括重油部分氧化重整制氢、天然气水蒸气重整制氢和煤气化制氢等。这些方法规模大,但能效及经济效应不高。电解水也可以制氢,该方法原料丰富,设备及操作简单,但是高能耗高成本,火力来源的发电途径也会造成污染物排放。太阳能耦联水解制氢,是实现碳中性的环保制氢方法,但目前由于金属有机类光能收集基团的能量转换效率偏低,有效催化媒介需贵重金属等问题,生产成本高昂。此外还有光合生物制氢及微生物发酵生物制氢[1]等绿色产氢途径,但同样由于经济效率或成本,这一类技术也没有实现工业生产。因此,廉价绿色氢能的规模化应用仍任重道远,需要跨越包括生物、光电物理、新型材料等领域的一系列科学技术的障碍才能实现。其中,对氢化酶的结构和催化机理的认识是解决包括光电制氢的廉价化学催化模拟物、高效氢电电池等重要的氢能开发或应用的基础生物技术研究之一。 1 氢化酶概述
1931年,Stephenson和Strickland首次发现并命名了氢化酶[2]。后来的研究表明它是催化电子传递反应链末端将质子还原产氢的含金属蛋白,其催化质子获电子产氢的可逆反应为:
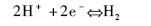
该反应为可逆催化反应,在有氢气和电子受体存在时,表现为吸氢反应;当有低电势的电子供体存在时,它可以利用水中的质子作为电子受体进行放氢反应。因此可以根据其电子供体和受体特异性对氢化酶进行分类。例如,Mortenson等[3]发现了铁氧化还原蛋白(Fd),它是一种来自于Clostridium pasteurianum的小的低氧化还原电势的铁硫蛋白,并且证明了它是梭菌属氢化酶的天然电子载体(反应1)。这种酶现在被IUBMB(International Union of Biochemistry and Molecular Biology,http://www.chem.qmul.ac.uk/iubmb/)记录为铁氧还蛋白氢化酶(Fd hydrogenase,EC 1.12. 7.2)。
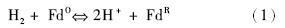
除Fd外,还有NAD (EC 1.12.1 .2 ),NADP,细胞色素 (EC 1.12.2.1),辅酶 F420(EC 1.12.98.1)等[4, 5]也可作为氢化酶的电子受体。此外,随着工业生物技术的发展,在(无细胞)体外酶机器催化系统的研究中,也产生了多种氢化酶的人工电子受体,它们的生产成本低廉,效率与天然电子受体相当,有着非常广阔的应用前景[6, 7]。表 1列出了被IUBMB记录的使用不同电子受体的各类氢化酶[5]。
| EC number | Name of the enzyme* |
| EC 1.12.1 .2 | NAD-linked hydrogenase or Hydrogen dehydrogenase |
| EC 1.12.1.3 | NADP+-reducing hydrogenase or Hydrogen dehydrogenase (NADP+) |
| EC 1.12.1.4 | Bifurcating [FeFe] hydrogenase or Hydrogenase (NAD+,ferredoxin) |
| EC 1.12.1.5 | Hydrogen dehydrogenase [NAD(P)+] or Hydrogenase II (ambiguous) |
| EC 1.12.2.1 | Cytochrome-c3 hydrogenase |
| EC 1.12.5.1 | Quinone-reactive Ni/Fe-hydrogenase or Hydrogen: quinone oxidoreductase |
| EC 1.12.7.2 | Ferredoxin hydrogenase |
| EC 1.12.98.1 | F420-reducing hydrogenase or Coenzyme F420 hydrogenase |
| EC 1.12.98.2 | 5,10-Methenyltetrahydromethanopterin hydrogenase or H2-forming 5,10-methylenetetrahydromethanopterin dehydrogenase |
| EC 1.12.98.3 | Methanosarcina-phenazine hydrogenase |
| EC 1.12.98.4 | Sulfhydrogenase or Sulfur reductase |
| EC 1.12.99.6 | Hydrogenase (acceptor) or Hydrogen: acceptor oxidoreductase |
| *Hydrogenase is named,in principle,as an enzyme catalyzing the uptake of H2 to reduce its electron acceptor,even if the evolution of H2 is relevantin vivo. The physiological electron acceptor of the enzyme can be read by the name of the enzyme | |
氢化酶常见的分类方法是依据其活性中心的金属成分分为三种类别:单铁氢化酶([Fe]-hydrogenase),双铁氢化酶([FeFe]-hydrogenase)及镍铁氢化酶([NiFe]-hydrogenase),这种分类方法在氢化酶的分子进化研究中也可以找到依据[4]。常见氢化酶为双铁氢化酶或镍铁氢化酶,而单铁氢化酶与前两者的结构和功能差异较大。
单铁氢化酶只在一部分产甲烷古细菌中被发现,目前研究较广泛的单铁氢化酶来自于Methanothermobacter marburgensis,简称Hmd[8]。它催化methenyl-H4MPT+和H2生成methylene-H4MPT和H+的可逆反应,该反应是CO2和H2转化为甲烷的中间过程[9, 10]。Hmd是由两个相同的亚基构成的同型二聚体,催化中心含有两个铁原子,但是不含硫铁簇[8, 10]。与双铁和镍铁氢化酶相比,Hmd不仅一级和三级结构上与它们存在明显差异,该酶活性中心所含的铁原子也不具有氧化还原活性,电子传递靠含铁的辅酶完成[11, 12]。
双铁氢化酶普遍存在于厌氧的原核生物及一些低等的真核生物,是唯一存在于真核生物中的氢化酶类型,位于细胞器叶绿体或者氢化酶体中[4]。严格厌氧条件下,该酶优先催化放氢反应[13]。多数双铁氢化酶都是仅由催化亚基组成的单体酶,也有二聚体、三聚体甚至四聚体[14, 15, 16],它们通常由含有活性中心(氢簇)的区域和包含铁硫簇的额外区域所组成。氢簇结构通常由一个双核中心[2Fe2S]及经半胱氨酸连接的一个 [4Fe-4S]簇组成,双核中心上的铁原子分别共价连接CO和CN-配体(图 1A),此外,铁原子还通过二硫配体与一个HN-(CH2-S-)2(二甲基或三甲基胺)连接[14]。在D. desulfuricans ATCC 7757[17]和Clostridium pasteurianum[14, 18]中的双铁氢化酶中都检测到了从分子表面到活性中心的单一疏水通道,但是分子动力学表明,氢气可以通过多种途径进入酶内部[19]。双铁氢化酶常为单体,活性中心结构相对简单,而其催化产氢活性很高,它们在氢产量上表现出的活性比[NiFe]氢化酶高出100倍以上[20];但该类酶空气中极易被氧化并不可逆失活,大大限制了它们的生物技术应用。
镍铁氢化酶广泛存在于细菌和古生菌中,常催化氢气的氧化反应兼其逆反应[21]。其核心结构是一个αβ异质二聚体,大亚基(α亚基)包含双金属活性位点,小亚基(β亚基)包含铁硫簇,大小亚基形成球状的异质二聚体。双金属镍铁中心深埋在大亚基内,通过四个半胱氨酸与酶蛋白相连,其结构与双铁氢化酶类似(图 1B)。另有镍铁硒氢化酶([NiFeSe]-hydrogenase),是镍铁氢化酶中的一种,其酶活中心一个与镍原子相连的半胱氨酸由硒代半胱氨酸(Selenocysteine,Sec)代替。铁原子有三个非蛋白配体,一个CO和两个CN-[22]。小亚基包含三个线性排列的铁硫簇,通常为[4Fe-4S]型的立方体(有的中间的铁硫簇为[3Fe-4S]型,如普通Desulfovibrio的镍铁氢化酶,具有较高的氧化还原电势),可以在氢气激活中心与氢化酶天然电子受体(或供体)之间导电。离活性中心最近的[4Fe-4S]簇是氢气激活所必需的[23, 24]。连通活性中心与分子表面的疏水通道可以促进气体进入活性中心[23, 25]。镍铁氢化酶结构较为复杂,其活性中心包埋较深,催化产氢活性较双铁氢化酶差,但其活性中心不易被氧化失活,对氧的敏感度相对较低,对其深入研究是制备有氧环境下生物产氢催化剂的重要参考依据。镍铁氢化酶活性中心的形成需要 将Ni,Fe,CO,和 CN-等基团在翻译后插入大小亚基间,而且,尽管不同生物体有拥有相似的成熟机制,但是各自的成熟系统却对其结构蛋白有高度特异性,因而使得氢化酶难以在异源宿主中被有效加工成熟[4]。

|
| 图 1 双铁和镍铁氢化酶活性中心的结构示意图 Fig. 1 The structure of the active center of [FeFe]-hydrogenases and [NiFe]-hydrogenases(A ) [FeFe]-hydrogenases; x maybe CH2,NH or O (B)[NiFe(Se)]-hydrogenases; (X) is one of H2O,HO2-,H-,H2,CO,OH- |
由于氢化酶结构及翻译后加工的复杂性,对氧化还原电位的高敏感性等问题,获取高纯活性蛋白一直是氢化酶分子机理研究的最大障碍,而解决这一技术障碍的有效办法是进行氢化酶的重组表达。随着近年来对氢化酶加工途径的初步了解,已经实现了越来越多氢化酶的重组表达,这些研究又促进了对氢化酶的催化及抗氧化分子机制的深入了解。而伴随着蛋白质工程、合成生物学及信息科学的进一步发展,氢化酶的酶工程研究也将进入新的时代,下面本文就近年来研究人员在氢化酶的同源和异源重组表达方面的工作进展作简要分析。 2.1 重组氢化酶的同源表达
氢化酶同源重组的报道不多,但这项研究使得研究人员可以较容易地获取有催化活性的全酶。最早的尝试来自Friedrich实验组,他们将耐氧的Ralstonia eutropha中编码膜结合氢化酶的结构基因及成熟加工的操作子构建到大的穿梭质粒中,成功地进行了该氢化酶的重组生产[26]。同源重组的一大优势是可以在氢化酶的亚基上加装用于纯化的亲合肽,如Lenz等将Strep Tag II的多肽利用同源重组表达生产了来自R. eutropha的可溶氢化酶,并因此对提纯的活性氢化酶进行了催化性能的分析[27]。来自耐热古细菌Pyrococcus furiosus的可溶氢化酶拥有四个催化亚基,其中两个亚基起酶蛋白结构稳定及电子传递功能,另两个起催化功能的亚基由Pf0893和Pf0894编码。Hopkins等使用了古菌自身的强启动子Pslp,完成了这两个催化亚基的同源重组表达,并发现该双聚体氢化酶可以利用偶联氧化还原反应中的产生的电子供体ferrodoxin[28]。随后,四亚基的可溶氢化酶及多亚基的膜结合氢化酶也均成功地进行了同源表达,但采取的过表达策略是将强表达启动子Pslp同源整合到目的基因的前端[29, 30, 31]。 2.2 重组氢化酶的异源表达
随着基因组学、分子遗传学、生物信息学等的发展,氢化酶的基因信息越来越明确,氢化酶在异源宿主菌株中成功进行功能性表达的研究成果越来越多[4, 32]。 2.2.1 双铁氢化酶异源重组表达
到目前为止,来自于丙酮丁醇梭菌(Clostridium acetobutylicum),丁酸梭菌(Clostridium butyricum),C. pasteurianum和莱茵衣藻(C. reinhardtii)的双铁氢化酶均已在异源菌株中表达,这些异源菌株包括大肠杆菌(Escherichia coli)、Shewanella oneidensis[33]、C. acetobutylicum、 Synechococcus sp. PCC7942[34]和Synechocystis sp. PCC6803[35]。
最初在异源菌株中功能性表达双铁氢化酶的尝试并不成功,因为缺乏对双铁氢化酶的成熟和装配过程的了解,宿主细胞不能合成完整的氢簇,所获重组氢化酶缺乏活性或酶活极低[34, 36, 37],但这些研究对揭示双铁氢化酶的成熟机制以及其表达的必需辅因子具有重要意义。2004年,Posewitz和团队在大肠杆菌(E. coli Bl-21 (DE3))中共表达了莱茵衣藻(C. reinhardtii) hydEF,hydG和hydA1基因,对重组表达的莱茵衣藻双铁氢化酶单体(C. reinhardtii HydA1)进行了纯化和酶活鉴定[38]。尽管C. reinhardtii HydA1在大肠杆菌中的表达水平并不高,但是该研究证明了辅助蛋白(HydE,HydF和 HydG)对于催化活性位点中的氢簇的生物合成和蛋白成熟是必需的。后续的研究中优化了功能性双铁氢化酶的表达系统,用共表达丙酮丁醇梭菌(C. acetobutylicum)的hydE,hydF和 hydG基因来代替莱茵衣藻(C. reinhardtii)的基因。King等在大肠杆菌中共表达C. acetobutylicum的hydE,hydF和 hydG以及C. reinhardtii,C. pasteurianum和C. acetobutylicum的双铁氢化酶单体基因,结果不但可以得到稳定表达活性,氢化酶的表达量也得到了大大提高[39]。2008年,Sybirna等发现C. reinhardtii的双铁氢化酶可以在希瓦氏菌(Shewanella oneidensis)中进行较高水平的功能性表达[33]。S. oneidensis是一种兼性的革兰氏阴性菌,很容易生长并进行遗传操作,与大肠杆菌不同,这株菌自身的双铁氢化酶与C. reinhardtii的双铁氢化酶有相似的密码子偏性。因此,它自身的成熟系统可以帮助CrHydA1的异源表达得到更高的酶产量(0.4~0.5 mg/L)和特异性酶活(740 μmol H2/(mgydmin)),比在C. acetobutylicum中的表达量(0.1mg/L)[40]以及在大肠杆菌系统中的特异性酶活(313 μmol H2/(mgydmin))[39]高出许多。
此外,蓝细菌(cyanobacterium)被用于双铁氢化酶的外源表达。Y. Asada等将Clostridium pasteurianum的双铁氢化酶克隆到Cyanobacterium Synechocystis PCC 7942(其自身含有镍铁氢化酶)进行异源表达,发现氢化酶活性增加,说明蓝细菌镍铁氢化酶的成熟机制似乎可以帮助双铁氢化酶的生物合成。2011年,Paola B.等使用蓝细菌Synechocystis sp.PCC 6803表达了有活性的C. reinhardtii的双铁氢化酶,也证明了蓝细菌可以在无双铁氢化酶合成辅助蛋白存在时,合成并正确折叠C. reinhardtii的双铁氢化酶[35](表 2)。然而,蓝细菌在没有双铁氢化酶合成辅助蛋白共表达时进行双铁氢化酶的折叠,这被认为是一种例外[34]。
| Hydrogenase | Expression host | Maturation protein | Whole cell activity | Purified enzyme activity | References |
| CrhydA1 | C. acetobutylicum | C. acetobutylicum | NR | 760c | [40] |
| CahydA | E. coli | C. acetobutylicum | 96a | 75c | [39] |
| CrhydA1 | E. coli | C. acetobutylicum | 61a | 150c | [39] |
| CshydA | in vitro | C. acetobutylicum | NA | 2.5c | [41] |
| CahydA | E. coli ΔiscR | C. acetobutylicum | NR | 96c | [42] |
| CphydA | in vitro | S. oneidensis | NA | 242c | [43] |
| CrhydA1 | in vitro | S. oneidensis | NA | 420c | [43] |
| CrhydA1 | S. oneidensis | S. oneidensis | 830a | 740c | [33] |
| CrhydA1 | Synechocystis sp. PCC 6803 | Synechocystis sp. PCC 6803 | 130b | NR | [35] |
| NA: Not applicable; NR: Not reported a Nmol H2-evolved/(min·ml-culture) from sodium dithionite-reduced methyl viologen b Nmol H2-evolved/(min·mg-chl) from sodium dithionite-reduced methyl viologen c μmol H2-evolved/(min·mg-protein) by sodium dithionite-reduced methyl viologen | |||||
藻类中表达重组氢化酶经常是为了生物产氢反应的需要,这面临更大的挑战[44],不仅需要克服细胞壁的限制将靶基因转化至叶绿体,还需要通过酶工程获得耐氧性能提升的双铁氢化酶,因此很多研究都集中在使用光合模式生物如莱茵衣藻来重组表达耐受双铁氢化酶,从而将光合生物制氢和水的氧化反应耦合。通过优化莱茵衣藻的细胞核转化方法,天然以及工程改造双铁氢化酶的重组表达最近取得了一定进展[45, 46],实验发现,通过球磨法、细胞壁破坏以及基因枪法进行莱茵衣藻的叶绿体转化是可行的[45, 47, 48]。而在藻类中进行可控制的氢化酶表达,达到光驱动产氢反应与氢化酶翻译后加工的协调一致,仍需要进一步研究。 2.2.2 镍铁氢化酶异源重组表达
由于镍铁氢化酶的重要性,对它们进行重组表达探索已久。然而,由于镍铁氢化酶结构复杂,结构基因和辅助基因复杂多样(表 3[49]),成熟机制的复杂性及宿主专一性等原因,获得功能性重组镍铁氢化酶的工作较双铁氢化酶更为困难,许多重组镍铁氢化酶没有生物活性。早在1987年,来自Desulfovibrio vulgaris的胞质可溶性镍铁氢化酶就在大肠杆菌中得到异源表达,但只产生了apo-enzymes,因活性中心缺失部分铁硫簇而没有催化活性[50]。随后,Acetomicrobium flavidum[51]和Rhodococcus opacus[52]中的镍铁氢化酶也在大肠杆菌进行了表达,但都没有氢化酶活性。
| Function | Species | |||||||||
| Escherichia coli | Ralstonia eutropha | Rhodobaoter capsulatus | Desulfovibrio | Methanosarcina | ||||||
| gigas | fructosovorans | barkeri | mazei | |||||||
| Hyd1 | Hyd2 | Hyd3 | MBH | SH | ||||||
| Histidine kinase | hoxJ | hoxJ | hupT | |||||||
| Sensor/SSU homologue | hoxB | hoxB | hupU | |||||||
| Sensor/LSU homologue | hoxC | hoxC | hupV | |||||||
| Response regulator (NtrC) | hydG | hoxA | hoxA | hupR | ||||||
| [2Fe-2S],NAD and FMN binding | hoxF | |||||||||
| [4Fe-4S] binding | hoxU | |||||||||
| Small subunit (SSU) | hyaA | hybO | hycG | hoxK | hoxY | hupS | hynA | hynA | echC | vhoG |
| Large subunit (LSU) | hyaB | hybC | hycE | hoxG | hoxH | hupL | hynB | hynB | echE | vhoA |
| Cytochrome b | hyaC | hybB | hoxZ | hupC/hupM | vhoC | |||||
| C-terminal peptidase | hyaD | hybD | hycI | hoxM | hoxW | hupD | hynC | hynC | vhoD | |
| Ni incorporation/maturation | hoxL | hupF | ||||||||
| SSU maturation? | hyaE | hoxO | hupG | |||||||
| SSU maturation | hyaF | hoxQ | hupH | |||||||
| O2 protection | hoxR | hupJ(5′) | ||||||||
| Electron transport | hybE | hoxT | hupJ(3′) | |||||||
| Fe(CN)2CO binding? | hoxV | hupK | ||||||||
| Ni incorporation/maturation | hybF | hybF | hypA | hypA1 | hypA2 | hypA | ||||
| Nickelin/Ni insertion | hypB | hypB | hypB | hypB1 | hypB2 | hypB | ||||
| Chaperone/maturation | hypC/hybG | hybG | hypC | hypC | hypC | hypC | hynD | |||
| Ni incorporation/maturation | hypD | hypD | hypD | hypD | hypD | hypD | ||||
| Purine derivative binding? | hypE | hypE | hypE | hypE | hypE | hypE | ||||
| CN/CO delivery | hypF | hypF | hypF | hypF1 | hypF2 | hypF | ||||
| CN/CO/acyl carrier? | hypX | hypX | ||||||||
| Transcriptonal activator | fhlA | |||||||||
| Repressor | hycA | |||||||||
| Iron sulfur protein | hycB | |||||||||
| Membrane protein | hycC | echB | ||||||||
| Membrane protein | hycD | echA | ||||||||
| Iron sulfur protein | hycF | echF | ||||||||
| Maturation | hycH | |||||||||
为此,人们首先选择将同源性较高的氢化酶在亲缘较近的宿主中进行异源重组。比如,巨大脱硫弧菌(Desulfovibrio gigas)镍铁氢化酶与Desulfovibrio fructosovorans氢化酶的氨基酸序列有64%一致,80%同源。Rousset等将D. gigas氢化酶的操纵子基因导入Desulfovibrio fructosovorans MR400中,由此得到了有活性的重组酶,但是重组酶的特异性酶活(0.16 U/mg)比原生酶酶活(1.0 U/mg)低很多,这可能是因为动力学因素的限制[53]。随后,混浊红球菌(Rhodococcus opacus,一株革兰氏阳性菌)的胞质镍铁氢化酶(SH)在罗氏真氧产碱杆菌(Ralstonia eutropha一株革兰氏阴性菌)中进行了表达,重组酶活近似达到了罗氏菌本身酶活的30%,尽管这两株菌种性差异性较大,但两株菌的SH相似性较高,四个亚基(hoxFUYH)有71%-86%的氨基酸一致性[54]。另有报道,Hydrogenovibrio marinus的镍铁氢化酶(HmMBH) 的两个催化亚基有和大肠杆菌的镍铁氢化酶1对应的催化亚基分别有73%(大亚基)和78%(小亚基)的同源性。HmMBH在大肠杆菌中功能性表达,该重组酶的产氢活性比大肠杆菌氢化酶1提高1.6-1.7倍,空气中纯化后的体外放氢活性也达到了28.8 nmol氢气/(minydmg蛋白)[55]。以上研究表明,亲缘较近的宿主细胞进行高同源性氢化酶的重组表达成功可能性较高。
实现镍铁氢化酶功能性表达的另一有效途径是进行氢化酶辅助蛋白的共表达,将包括结构基因、辅助基因、调控基因等在内的相关基因全部转入宿主细胞。Lenz等将Ralstonia eutropha的膜结合镍铁氢化酶(MBH)操纵子(共21个基因)全部构建在广泛宿主载体中,通过接合作用导入没有氢化酶的宿主菌株施氏假单胞菌(Pseudomonas stutzeri)中,结果该异源表达系统在无氢化酶的宿主菌P. stutzeri中产生了镍铁氢化酶活性。该实验同时证明,R. eutropha MBH的所有操纵子基因,包括结构基因(hoxKGZ),MBH特异性辅助基因(hoxMLOQRTV),七个hyp基因(hypABFCDEX),和调控基因(hoxABCJ)都是外源重组表达所必需的[26]。有些情况下,用于异源表达的宿主细胞自身所带的氢化酶成熟因子可以协助完成外源氢化酶的加工,因此只需共表达部分辅助因子即可实现目的蛋白的功能性表达,如Sun等利用大肠杆菌氢化酶缺陷菌株表达了一株极端嗜热古生菌Pyrococcus furiosus (Pf)的胞质镍铁氢化酶(SHI),他们在大肠杆菌中用四个载体共表达Pf的十三个基因(PfSHI的四个结构基因与9个成熟蛋白基因),成功实现了PfSHI的功能性表达;为了确定在大肠杆菌中成功表达活性PfSHI所需的最少基因数,研究者们进行了许多后续实验,发现在大肠杆菌中只需将PfSHI的四个结构基因(PF0891-PF0894)与一个成熟蛋白的基因(frxA),即可实现PfSHI的功能性表达[56]。这说明,至少部分情况下,大肠杆菌可以利用其自身的镍铁氢化酶成熟系统帮助异源氢化酶进行翻译后加工。
随着合成生物学的发展,高水平表达镍铁氢化酶越来越可能实现。2013年,Schiffels等用一种新型克隆平台实现了Cupriavidus necator H16(原Ralstonia eutropha H16)的胞质镍铁氢化酶(SH)在大肠杆菌中的高水平表达。该平台每个基因都有自己的启动子和终止子,它们被分别构建到一系列相容性质粒上。用该平台在大肠杆菌中共表达SH结构基因hoxFUYHI以及一套专用的辅助基因(七个hyp基因(hypC1D1E1A2B2F2X)和hoxW(编码一个特异性肽链内切酶),结果纯化出的功能性SH的产量达到27 mg/L发酵液,特异性酶活达到了7.2±1.15 U/mg,在厌氧下进一步提纯,比酶活高达230 U/mg[57]。该平台为氢化酶的生物合成提供了新的方向。尽管镍铁氢化酶的异源重组研究有了长足进步,但多数的重组酶和原生氢化酶相比,比酶活都严重下降(表 4),这表明外源宿主细胞对重组氢化酶的成熟加工效率依然不高,因此,对氢化酶的翻译后加工机理需要进一步的探究和认识。
| Hydrogenase | Expression host | Maturation protein | Whole cell activity | Purified enzyme activity | References |
| D. gigas | D. fructosovorans | D. gigas | 0.16a | NR | [49] |
| R. opacus | R. eutropha | R. eutropha | 7.75b | NR | [54] |
| R. eutropha | P. stutzeri | R. eutropha | 19c | NR | [26] |
| P. furiosus | E. coli | P. furiosus | 2.9d | 100 | [56] |
| Alteromonas macleodii | Synechococcus elongatus | Alteromonas macleodii | 7×10-5d | NR | [58] |
| Synechocystis sp. PCC 6803 | E. coli | Synechocystis sp. PCC 6803 | 0.04d | NR | [59] |
| NR: Not reported a μmol H2-consumed/(min·mg) for reduction of NAD+ b μmol H2-consumed/(min·mg) for reduction of benzyl viologen c μmol H2-consumed/(min·mg) for reduction of methylene blue d μmol H2-evolved/(min·mg-protein) by sodium dithionite-reduced methyl viologen | |||||
至此,氢化酶的重组表达研究还远远不够,因为理想的重组氢化酶不仅要有很高的耐氧性能、极强的催化稳定性能,还要有可以利用廉价的电子供体等天然氢化酶所没有的酶学特点,而目前多数重组酶尚没有达到天然酶的催化性能;此外,重组研究的另一重要方向是促进产生最小化的人工氢化酶蛋白体,从而为氢化酶化学模拟催化剂的开发提供原酶模型,这项研究工作需要对氢化酶的分子催化机理和耐氧机制及成熟机理有进一步的认识,并结合氢化酶的高通量筛选技术,运用生物信息学、合成生物学的技术手段,且经不断努力才能实现。
| [1] | Ihara M, Nakamoto H, Kamachi T, et al. Photoinduced hydrogen production by direct electron transfer from photosystem I cross-linked with cytochrome c(3) to NiFe -hydrogenase. Photochemistry and Photobiology, 2006, 82(6): 1677-1685. |
| [2] | Stephenson M, Stickland L H. Hydrogenase: A bacterial enzyme activating molecular hydrogen. I. The properties of the enzyme. Biochemical Journal, 1931, 25(1): 205-214. |
| [3] | Mortenson L E, Carnahan J E, Valentine R C. An electron transport factor from clostridium pasteurianum. Biochemical and Biophysical Research Communications, 1962, 7(6): 448-452. |
| [4] | Vignais P M, Billoud B. Occurrence, classification, and biological function of hydrogenases: an overview. Chemical Reviews, 2007, 107(10): 4206-4272. |
| [5] | Yagi T, Higuchi Y. Studies on hydrogenase. Proceedings of the Japan Academy, Series B, 2013, 89(1): 16-33. |
| [6] | Paul C E, Arends I, Hollmann F. Is simpler Better? Synthetic nicotinamide cofactor analogues for redox chemistry. Acs Catalysis, 2014, 4(3): 788-797. |
| [7] | Rollin J A, Tam T K, Zhang Y H P. New biotechnology paradigm: cell-free biosystems for biomanufacturing. Green Chemistry, 2013, 15(7): 1708-1719. |
| [8] | Zirngibl C, Vandongen W, Schworer B, et al. H-2-forming methylenetetrahydromethanopterin dehydrogenase, a novel type of hydrogenase without iron-sulfur clusters in methanogenic archaea. European Journal of Biochemistry, 1992, 208(2): 511-520. |
| [9] | Thauer R K. Biochemistry of methanogenesis: a tribute to Marjory Stephenson. Microbiology-Uk, 1998, 144: 2377-2406. |
| [10] | Thauer R K, Klein A R, Hartmann G C. Reactions with molecular hydrogen in microorganisms: evidence for a purely organic hydrogenation catalyst. Chemical Reviews, 1996, 96(7): 3031-3042. |
| [11] | Korbas M, Vogt S, Meyer-Klaucke W, et al. The iron-sulfur cluster-free hydrogenase (Hmd) is a metalloenzyme with a novel iron binding motif. Journal of Biological Chemistry, 2006, 281(41): 30804-30813. |
| [12] | Shima S, Lyon E J, Sordel-Klippert M S, et al. The cofactor of the iron-sulfur cluster free hydrogenase Hmd: Structure of the light-inactivation product. Angewandte Chemie-International Edition, 2004, 43(19): 2547-2551. |
| [13] | Friedrich B, Fritsch J, Lenz O. Oxygen-tolerant hydrogenases in hydrogen-based technologies. Current Opinion in Biotechnology, 2011, 22(3): 358-364. |
| [14] | Nicolet Y, Cavazza C, Fontecilla-Camps J C. Fe-only hydrogenases: structure, function and evolution. Journal of Inorganic Biochemistry, 2002, 91(1): 1-8. |
| [15] | Peters J W. Structure and mechanism of iron-only hydrogenases. Current Opinion in Structural Biology, 1999, 9(6): 670-676. |
| [16] | Vignais P M, Billoud B, Meyer J. Classification and phylogeny of hydrogenases. Fems Microbiology Reviews, 2001, 25(4): 455-501. |
| [17] | Nicolet Y, Piras C, Legrand P, et al. Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an active site Fe binuclear center. Structure with Folding & Design, 1999, 7(1): 13-23. |
| [18] | Peters J W, Lanzilotta W N, Lemon B J, et al. X-ray crystal structure of the Fe-only hydrogenase (Cpl) from Clostridium pasteurianum to 1.8 angstrom resolution. Science, 1998, 282(5395): 1853-1858. |
| [19] | Cohen J, Kim K, King P, et al. Finding gas diffusion pathways in proteins: application to O-2 and H-2 transport in Cpl FeFe -hydrogenase and the role of packing defects. Structure, 2005, 13(9): 1321-1329. |
| [20] | Frey M. Hydrogenases: Hydrogen-activating enzymes. Chembiochem, 2002, 3(2-3): 153-160. |
| [21] | Kim J Y H, Cha H J. Recent progress in hydrogenase and its biotechnological application for viable hydrogen technology. Korean Journal of Chemical Engineering, 2013, 30(1): 1-10. |
| [22] | Pierik A J, Roseboom W, Happe R P, et al. Carbon monoxide and cyanide as intrinsic ligands to iron in the active site of NiFe -hydrogenases - NiFe(CN)2CO, biology's way to activate H2. Journal of Biological Chemistry, 1999, 274(6): 3331-3337. |
| [23] | Fontecilla-Camps J C, Frey M, Garcin E, et al. Hydrogenase: A hydrogen-metabolizing enzyme. What do the crystal structures tell us about its mode of action? Biochimie, 1997, 79(11): 661-666. |
| [24] | Volbeda A, Charon M H, Piras C, et al. Crystal-structure of the nickel-iron hydrogenase from desulfovibrio-gigas. Nature, 1995, 373(6515): 580-587. |
| [25] | Montet Y, Amara P, Volbeda A, et al. Gas access to the active site of Ni-Fe hydrogenases probed by X-ray crystallography and molecular dynamics. Nature Structural Biology, 1997, 4(7): 523-526. |
| [26] | Lenz O, Gleiche A, Strack A, et al. Requirements for heterologous production of a complex metalloenzyme: the membrane-bound NiFe hydrogenase. Journal of bacteriology, 2005, 187(18): 6590-6595. |
| [27] | Lauterbach L, Lenz O. Catalytic production of hydrogen peroxide and water by oxygen-tolerant NiFe -hydrogenase during H-2 cycling in the presence of O-2. Journal of the American Chemical Society, 2013, 135(47): 17897-17905. |
| [28] | Hopkins R C, Sun J S, Jenney F E, et al. Homologous expression of a subcomplex of pyrococcus furiosus hydrogenase that interacts with pyruvate ferredoxin oxidoreductase. PloS one, 2011, 6(10). |
| [29] | Chandrayan S K, McTernan P M, Hopkins R C, et al. Engineering hyperthermophilic archaeon Pyrococcus furiosus to overproduce its cytoplasmic NiFe -hydrogenase. Journal of Biological Chemistry, 2012, 287(5): 3257-3264. |
| [30] | McTernan P M, Chandrayan S K, Wu C H, et al. Engineering the respiratory membrane-bound hydrogenase of the hyperthermophilic archaeon Pyrococcus furiosus and characterization of the catalytically active cytoplasmic subcomplex. Protein Engineering, Design & Selection: PEDS, 2015, 28(1): 1-8. |
| [31] | McTernan P M, Chandrayan S K, Wu C H,et al. Intact functional fourteen-subunit respiratory membrane-bound NiFe -hydrogenase complex of the hyperthermophilic archaeon Pyrococcus furiosus. Journal of Biological Chemistry, 2014, 289(28): 19364-19372. |
| [32] | English C M, Eckert C, Brown K, et al. Recombinant and in vitro expression systems for hydrogenases: new frontiers in basic and applied studies for biological and synthetic H2 production. Dalton transactions, 2009(45): 9970-9978. |
| [33] | Sybirna K, Antoine T, Lindberg P, et al. Shewanella oneidensis: a new and efficient system for expression and maturation of heterologous Fe-Fe hydrogenase from Chlamydomonas reinhardtii. BMC Biotechnology, 2008, 8:73. |
| [34] | Asada Y, Koike Y, Schnackenberg J, et al. Heterologous expression of clostridial hydrogenase in the cyanobacterium Synechococcus PCC7942. Biochimica Et Biophysica Acta-Gene Structure and Expression, 2000, 1490(3): 269-278. |
| [35] | Berto P, D'Adamo S, Bergantino E, et al. The cyanobacterium Synechocystis sp PCC 6803 is able to express an active FeFe -hydrogenase without additional maturation proteins. Biochemical and Biophysical Research Communications, 2011, 405(4): 678-683. |
| [36] | Atta M, Meyer J. Characterization of the gene encoding the Fe -hydrogenase from Megasphaera elsdenii. Biochimica Et Biophysica Acta-Protein Structure and Molecular Enzymology, 2000, 1476(2): 368-371. |
| [37] | Gorwa M F, Croux C, Soucaille P. Molecular characterization and transcriptional analysis of the putative hydrogenase gene of Clostridium acetobutylicum ATCC 824. Journal of Bacteriology, 1996, 178(9): 2668-2675. |
| [38] | Posewitz M C, King P W, Smolinski S L, et al. Discovery of two novel radical S-adenosylmethionine proteins required for the assembly of an active Fe hydrogenase. Journal of Biological Chemistry, 2004, 279(24): 25711-25720. |
| [39] | King P W, Posewitz M C, Ghirardi M L, et al. Functional studies of [FeFe] hydrogenase maturation in an Escherichia coli biosynthetic system. Journal of Bacteriology, 2006, 188(6): 2163-2172. |
| [40] | Girbal L, von Abendroth G, Winkler M, et al. Homologous and heterologous overexpression in Clostridium acetobutylicum and characterization of purified clostridial and algal Fe-only hydrogenases with high specific activities. Applied and Environmental Microbiology, 2005, 71(5): 2777-2781. |
| [41] | McGlynn S E, Ruebush S S, Naumov A, et al. In vitro activation of FeFe hydrogenase: new insights into hydrogenase maturation. Journal of Biological Inorganic Chemistry, 2007, 12(4): 443-447. |
| [42] | Akhtar M K, Jones P R. Deletion of iscR stimulates recombinant clostridial Fe-Fe hydrogenase activity and H-2-accumulation in Escherichia coli BL21(DE3). Applied Microbiology and Biotechnology, 2008, 78(5): 853-862. |
| [43] | Boyer M E, Stapleton J A, Kuchenreuther J M, et al. Cell-free synthesis and maturation of FeFe hydrogenases. Biotechnology and Bioengineering, 2008, 99(1): 59-67. |
| [44] | Cohen J, Kim K, Posewitz M, et al. Molecular dynamics and experimental investigation of H-2 and O-2 diffusion in Fe -hydrogenase. Biochemical Society Transactions, 2005, 33: 80-82. |
| [45] | Kindle K L. High-frequency nuclear transformation of Chlamydomonas reinhardtii.PNAS, 1990, 87(3): 1228-1232. |
| [46] | Lumbreras V, Stevens D R, Purton S. Efficient foreign gene expression in Chlamydomonas reinhardtii mediated by an endogenous intron. Plant Journal, 1998, 14(4): 441-447. |
| [47] | Blowers A D, Bogorad L, Shark K B, et al. Studies on chlamydomonas chloroplast transformation - foreign dna can be stably maintained in the chromosome. Plant Cell, 1989, 1(1): 123-132. |
| [48] | Boynton J E, Gillham N W, Harris E H, et al. Chloroplast transformation in chlamydomonas with high-velocity microprojectiles. Science, 1988, 240(4858): 1534-1538. |
| [49] | Casalot L, Rousset M. Maturation of the NiFe hydrogenases. Trends in Microbiology, 2001, 9(5): 228-237. |
| [50] | Voordouw G, Hagen W R, Krusewolters K M, et al. Purification and characterization of Desulfovibrio-vulgaris (hildenborough) hydrogenase expressed in Escherichia-coli. European Journal of Biochemistry, 1987, 162(1): 31-36. |
| [51] | Mura G M, Pedroni P, Pratesi C, et al. The Ni-Fe hydrogenase from the thermophilic bacteriuna Acetomicrobium flavidum. Microbiology-UK, 1996, 142: 829-836. |
| [52] | Grzeszik C, Lubbers M, Reh M, et al. Genes encoding the NAD-reducing hydrogenase of Rhodococcus opacus MR11. Microbiology-UK, 1997, 143: 1271-1286. |
| [53] | Rousset M, Magro V, Forget N, et al. Heterologous expression of the Desulfovibrio gigas NiFe hydrogenase in Desulfovibrio fructosovorans MR400. Journal of Bacteriology, 1998, 180(18): 4982-4986. |
| [54] | Porthun A, Bernhard M, Friedrich B. Expression of a functional NAD-reducing [NiFe]hydrogenase from the gram-positive Rhodococcus opacus in the gram-negative Ralstonia eutropha. Archives of Microbiology, 2002, 177(2): 159-166. |
| [55] | Kim J Y, Jo B H, Cha H J. Production of biohydrogen by heterologous expression of oxygen-tolerant Hydrogenovibrio marinus [NiFe]-hydrogenase in Escherichia coli. Journal of Biotechnology, 2011, 155(3): 312-319. |
| [56] | Sun J, Hopkins R C, Jenney F E, et al. Heterologous expression and maturation of an NADP-dependent NiFe -hydrogenase: a key enzyme in biofuel production. PloS one, 2010, 5(5). |
| [57] | Schiffels J, Pinkenburg O, Schelden M, et al. An innovative cloning platform enables large-scale production and maturation of an oxygen-tolerant NiFe -hydrogenase from Cupriavidus necator in Escherichia coli. PloS one, 2013, 8(7). |
| [58] | Weyman P D, Vargas W A, Tong Y K, et al. Heterologous expression of Alteromonas macleodii and Thiocapsa roseopersicina NiFe hydrogenases in Synechococcus elongatus. PloS one, 2011, 6(5): 8. |
| [59] | Wells M A, Mercer J, Mott R A, et al. Engineering a non-native hydrogen production pathway into Escherichia coli via a cyanobacterial NiFe hydrogenase. Metabolic Engineering, 2011, 13(4): 445-453. |