 2021, Vol. 32
2021, Vol. 32扩展功能
文章信息
- 董昊炜, 南春燕, 周秋明, 袁浩, 李翔宇, 彭恒, 马雅军
- DONG Hao-wei, NAN Chun-yan, ZHOU Qiu-ming, YUAN Hao, LI Xiang-yu, PENG Heng, MA Ya-jun
- 基于16S rRNA高通量测序的现场中华按蚊及其孳生地水体菌群结构研究
- Composition of microbiota in Anopheles sinensis and water in larvae breeding site: A study based on 16S rRNA high-throughput sequencing
- 中国媒介生物学及控制杂志, 2021, 32(5): 533-540
- Chin J Vector Biol & Control, 2021, 32(5): 533-540
- 10.11853/j.issn.1003.8280.2021.05.005
-
文章历史
- 收稿日期: 2021-05-20




2 海军军医大学海军医学系, 上海 200433
2 College of Naval Medicine, Naval Medical University, Shanghai 200433, China
中华按蚊(Anopheles sinensis)是我国分布最广的按蚊种类,是所在地区,尤其是长江中下游平原地区的重要传疟媒介[1]。疟疾是严重危害人类健康的全球性传染病,在我国部分地区也曾严重流行,经多年的大力防治,从2017年起至今我国已经实现连续无本地感染病例报告[2]。2021年6月世界卫生组织确认中国已消除疟疾。然而,在疟疾消除后,我国仍然面临着疟疾流行的风险,主要威胁来自于输入性病例由本地媒介传播造成的继发性感染[3]。
众所周知,疟原虫在按蚊体内经历一系列复杂的发育和繁殖过程才可以感染人。蚊虫肠道作为相应病原体的首道防线,定植着大量微生物,包括浮游生物、细菌与真菌等。肠道共生菌在蚊虫的许多生理活动中扮演着重要的角色,如营养与代谢、发育与生殖、免疫和防御等[4]。多项研究显示,按蚊肠道共生菌可影响疟原虫的发育[5-7],如:有研究报道,沙雷菌(Serratia sp.)和肠杆菌(Enterobacter sp.)在按蚊中肠存在时,能抑制疟原虫的发育,显著降低其发育成卵囊的数量[8-10]。因此,开展按蚊细菌多样性和菌群结构的分析可为深入研究媒介与疟原虫相互关系提供理论基础,最终为疟疾防控提供新思路[4, 11-12]。
中华按蚊共生菌的分离和多样性研究报告不多[13-15],本课题组基于高通量测序技术对其幼蚊的肠道细菌宏基因组进行了初步分析[16],Gao等[17]比较了江苏、云南和辽宁现场的中华按蚊肠道共生菌群的结构组成,发现沙雷菌是肠道核心菌群(core gut microbiota)[18]。高通量测序技术分析菌群群落,无需分离培养即可获得覆盖度高的菌群分类信息,以此揭示被低估的共生菌群多样性。细菌16S rDNA是细菌系统分类学研究中最常用的标志,该基因片段包括9个可变区和10个保守区,保守区序列可反映物种间的亲缘关系,而可变区序列则能体现物种间的差异[19]。鉴于蚊虫共生细菌菌群与生活史阶段、生理状态以及环境等关系密切[20-23],本研究对现场采集的中华按蚊饱血和未吸血雌蚊、孳生地水样的细菌进行高通量测序,分析其菌群组成和多样性,并比较了其中的差异。
1 材料与方法 1.1 供试样本 1.1.1 蚊虫2012年8月,在上海市嘉定区华亭镇奶牛场,用吸蚊器人工捕捉中华按蚊雌蚊,麻醉后单蚊单管保存。
1.1.2 孳生地水样在奶牛场邻近的稻田,舀取有幼蚊或蛹活动的水,去除其中的植物残渣和活动的水生生物,分别装入预先灭菌的容器,带回实验室,4 ℃保存。
1.2 主要试剂蚊虫宏基因组提取试剂盒(MasterPureTM DNA Purification Kit)和水样宏基因组提取试剂盒(Meta-G-NomeTM DNA Purification Kit)均购自美国Epicentre公司,组织细胞基因组DNA提取试剂盒购自上海旭飞生物科技有限公司,引物合成和序列测定由上海铂尚生物技术有限公司完成,MiSeq宏基因组测序由上海派森诺生物科技有限公司完成,测序仪(Miseq System SY-410-1003)为美国Illumina公司产品。
1.3 方法 1.3.1 蚊种鉴定将雌蚊样本置于解剖镜下,观察其形态特征,鉴定为赫坎按蚊种团(An. hyrcanus group)的成员种[1]。对形态鉴定为该种团成员种的个体用75%乙醇溶液润洗3次后,用无菌的解剖针将头部与胸腹部分开,头部用于分子鉴别,胸腹部用于宏基因组菌群分析。单蚊头部基因组DNA抽提按照试剂盒说明进行,分子鉴别的方法参照文献[24]。
1.3.2 宏基因组抽提将鉴别为中华按蚊的雌蚊胸腹部分别合并,分为未吸血雌蚊(A)和饱血雌蚊(B)2组,为避免实验误差,每组分为2个样本,每个样本由10只合并,按照试剂盒说明抽提宏基因组。3幼虫孳生地水样(W)分为2个样本,宏基因组抽提操作按照试剂盒说明。
1.3.3 Miseq宏基因组测序以宏基因组为模板扩增细菌的16S rDNA-V4区片段,引物为[25]:正向5'-AYT GGG YDT AAA GNG-3',反向5'-TAC NVG GGT ATC TAA TCC-3'。PCR产物通过3'~5'核酸外切酶及聚合酶的共同作用,修复带有突出末端的DNA片段,在3'端引入单碱基“A-”,与3'端“T-”的接头连接,利用PCR选择性富集有接头的DNA片段,同时扩增DNA文库,逐步稀释定量后上机测序,按照Illumina公司标准流程进行。
1.3.4 生物信息学分析首先,对原始数据进行质量控制,舍弃低质量序列,用Flash(V1.0.3)软件进行双端拼接,对拼接的序列进行过滤,剔除连续相同碱基 > 6和模糊碱基N > 1的序列。随后,应用Qiime软件,在≥97.00%的相似度下,对获得的序列进行操作分类单元(operational taxonomic unit,OTU)聚类。通过比对亲缘关系的方法,注释每个OTU的分类学信息。利用Mothor软件计算基于菌群丰度的Alpha多样性指数,包括反映群落丰富性的Chao1指数、反映菌群多样性的香农指数(Shannon index)和辛普森指数(Simpson index),以及覆盖度(Good’s coverage)。在门、科和属水平分析单样本的细菌菌群组成和丰度,以及多样本的细菌菌群相对丰度,并进行t检验,P < 0.05为差异有统计学意义。另外,对测序数据进行基于群落比较的β多样性分析,包括主成分分析(principal component analysis,PCA)和热图聚类。
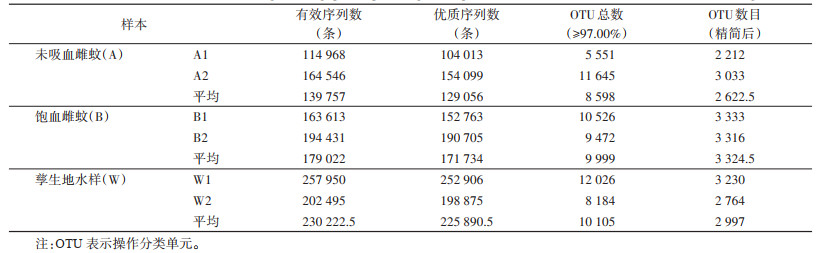
2 结果 2.1 测序序列与OTU统计本研究获得中华按蚊雌蚊(A和B)及其W 3组,共6个样本的16S rDNA-V4区片段,平均有效序列和优质序列数为139 757/129 056(A)、179 022/171 734(B)和230 222.5/225 890.5(W)。见表 1。
|
通过比对归并获得≥97.00%相似度OTU数目,W样本中最多,为10 105,其次是B和A样本,而精简后的OTUs数目,从多到少的顺序是B > W > A(表 1)。对精简后的OTU进行注释,隶属20个门,144个科,295个属。
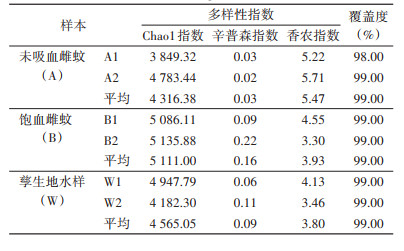
2.2 Alpha多样性分析Alpha多样性可用Chao1指数、辛普森指数和香农指数等数据进行分析,以评估样本中细菌群落的丰富度和多样性。从各组的平均数看,Chao1指数衡量本研究样本的细菌丰富度从高到低的顺序是B > W > A;辛普森指数显示物种多样性,数值越低物种多样性越高,该指数从高到低的顺序为B > W > A,物种多样性从高到低的顺序为A > W > B;而香农指数显示的顺序为A > B > W。见表 2。
|
依据覆盖度的数据,可见除样本A1(98.00%)外,其余5个样本的细菌群落采样覆盖率均接近100%(表 2)。稀疏曲线显示,总体的趋势是所有样本在20 000条序列之前,观察到的OTU随着序列数的增加而急剧上升,之后随着测序深度加深,上升平缓并接近饱和,到达平台期。
2.3 菌群群落结构分析本研究统计和分析菌群群落结构的样本包括A、B、W,以及该孳生地生活的幼蚊(L),数据参见文献[16],取组的平均值进行计算。
2.3.1 单样本细菌菌群丰度 2.3.1.1 门水平统计各样本检测到的细菌,其中部分种类未获得注释,在此列出丰度 > 1.00%的门。A样本中的细菌隶属17个门,变形菌门(Proteobacteria)所占比例最高(44.68%),其次为厚壁菌门(Firmicutes,28.83%)、拟杆菌门(Bacteroidetes,4.47%)和放线菌门(Actinobacteria,2.92%),其余13个门占19.10%;B样本中的细菌隶属19个门,变形菌门所占比例为73.92%,其次是厚壁菌门(16.48%)、拟杆菌门(2.49%)和放线菌门(2.22%),共占95.12%;L中的细菌隶属于20个门,其中变形菌门占71.22%,拟杆菌门、蓝细菌门(Cyanobacteria)和厚壁菌门分别占9.19%、3.72%和2.02%,其余16个门仅占13.85%;W样本中的细菌隶属于18个门,其中优势的包括厚壁菌门(37.84%)、拟杆菌门(32.80%)和变形菌门(24.33%),其余的15个门占5.03%。见图 1。

|
| 注:A表示未吸血雌蚊;B表示饱血雌蚊;L表示幼蚊;W表示孳生地水样。 图 1 中华按蚊与孳生地水样中的细菌在门水平的组成和丰度 Figure 1 Bacterial composition and abundance at the phylum level of Anopheles sinensis and water in larvae breeding site |
| |
统计各样本检测到的细菌,由于属的数量多,在此列出丰度≥10.00%的属。A样本中的细菌隶属于207个属,不动杆菌属(Acinetobacter)和泛菌属(Pantoea)分别占41.35%和10.58%;B样本中的细菌隶属254个属,Asaia、不动杆菌属和泛菌属分别占40.85%、25.71%和12.70%。L样本中的细菌隶属259个属,丰度≥10.00%的属有红细菌属(Rhodobacter,15.24%)、Thorsellia(13.57%)和嗜甲基菌属(Methylophilus,10.93%);W中的细菌隶属于244个属,包括Desulfosporosinus(24.19%)、弯杆菌属(Flectobacillus,19.48%)、产黄菌属(Flavobacterium,15.27%)、假单胞菌属(Pseudomonas,12.19%)和Alkalibacter(10.28%)。
2.3.2 多样本细菌物种分布 2.3.2.1 门水平以L1为基准,在20个门中选择8个样本共有的7个优势菌门,统计分析其相对丰度,可见在A1、A2、B1、B2、L1和L2中变形菌门所占比例均为最高,而W1与W2的优势门不同,分别为厚壁菌门和拟杆菌门。见图 2。

|
| 注:A表示未吸血雌蚊;B表示饱血雌蚊;L表示幼蚊;W表示孳生地水样。 图 2 中华按蚊各虫期与孳生地水体中优势细菌在门水平的组成和相对丰度 Figure 2 Bacterial composition and relative abundance at the phylum level of Anopheles sinensis adults and water in larvae breeding site |
| |
各样本间细菌菌群差异t检验的结果显示,差异性的门数分别为1个(B/A)、8个(L/B、L/A)和9个(W/A、W/B、W/L),A与B样本之间差异有统计学意义的门最少,而W与L、A、B之间的差异性较大。统计其中差异倍数≥10的门仅有3个,分别是L/A的OP10(18.18)、W/B的粘胶球形菌门(-21.10)和W/L的广古菌门(-14.75)。
2.3.2.2 科水平以L1为基准,选择共有的39个优势科比较其相对丰度(图 3),在此列出丰度≥1.00%的科。结果显示各组内样本间的重复性尚可,在A1和A2中莫拉菌科(Moraxellaceae)丰度均为最高;B1与B2的肠杆菌科(Enterobacteriaceae)与醋杆菌科(Acetobacteraceae)相对丰度排位略有差异,肠杆菌科与醋杆菌科在B1中为第1和第3,第2为莫拉菌科,而B2中醋杆菌科排首位,其次是肠杆菌科;L1和L2中所含产碱菌科(Alcaligenaceae)的相对丰度均为最高;在W1中丰度≥10.00%的是消化球菌科(Peptococcaceae)和优杆菌科(Eubacteriaceae),W2中为噬纤维菌科(Cytophagaceae)、消化球菌科和假单胞菌科(Pseudomonadaceae)。

|
| 注:A表示未吸血雌蚊;B表示饱血雌蚊;L表示幼蚊;W表示孳生地水样。 图 3 中华按蚊与孳生地水样中优势细菌在科水平的组成和相对丰度 Figure 3 Bacterial composition and relative abundance at the family level of Anopheles sinensis adults and water in larvae breeding site |
| |
Beta多样性是不同生态系统间多样性的比较,是物种组成沿环境梯度或在群落间的变化率,用来表示生物种类对环境异质性的反应。本研究8个样本的PCA结果显示,同组的样本,即A1与A2、B1与B2、L1与L2、W1与W2分别聚在一起,同组样本的细菌多样性相近,同是成蚊的A与B分别聚在一个区。见图 4。

|
| 注:A表示未吸血雌蚊;B表示饱血雌蚊;L表示幼蚊;W表示孳生地水样。 图 4 中华按蚊与孳生地水样的细菌主成分分析结果 Figure 4 Principal component analysis of bacteria in Anopheles sinensis and water in larvae breeding site |
| |
在属水平进行热图聚类分析可以反映样本间的相似性。聚类关系显示,同组的样本首先聚为一支,之后是W与L聚在一起,B与A聚为另一支,两者互为姐妹支,W与L、A与B样本中的细菌相似程度高。
3 讨论本研究基于16S rDNA高通量测序技术对上海市嘉定区的中华按蚊A和B样本,以及W样本的细菌宏基因组组成进行了多样性分析,并与其幼蚊进行了菌群结构的比较。应用细菌的16S rDNA-V4可变区序列,可以有效解析中华按蚊肠道和稻田水体的细菌种类[19, 25-26]。高通量测序不仅可以获得接近实际情况的蚊虫共生细菌类群,其细菌OTUs数远远大于应用变性梯度凝胶电泳法分离到的细菌种数[13-14],而且测序数据量还可以准确评估菌群的相对丰度。
理论上,蚊虫共生细菌群落的多样性从大到小应为孳生地水体 > 幼蚊,但本研究结果显示稻田水样中细菌丰富度指数小于幼蚊,分析原因可能是水中含有多种化学物质,如腐殖质酸、残留的杀虫剂和重金属离子等,降低了宏基因组提取和PCR扩增的质量,导致细菌丰富度受损。Wang等[20]应用高通量测序研究冈比亚按蚊(An. gambiae)生活史各期肠道微生物组成,发现在孳生地中变形菌门占据主导地位,其次是蓝细菌门。本研究的稻田水体中,厚壁菌门、拟杆菌门和变形菌门占优势地位,而蓝细菌门所占比例很少,可能是由于采集的水样在4 ℃冰箱中放置了一段时间后才进行宏基因组抽提和测序,其中光合作用的细菌,如蓝细菌门种类死亡,导致基因组降解而未获得检测。然而,在属水平进行热图聚类分析,仍然可以看出W样本中的细菌与其中孳生的幼蚊细菌相似程度较高。
已证实孳生地水体中细菌是幼蚊摄取的食物之一,但其肠道环境对其有一定的选择性,仅少数细菌才可定植,幼蚊发育到成蚊阶段时,经历了蛹期胎粪排除,新羽化的成蚊肠道几乎为无菌状态,因此,幼蚊与成蚊阶段细菌菌群组成差异较大。本研究的中华按蚊幼蚊与成蚊间差异显著的细菌有8个门,远远大于B与A样本间的1个门。实验室研究发现,雌蚊吸血后的肠道环境会趋向于选择有溶血能力的细菌,吸血时体温的迅速增高、氧胁迫压力的降低都让细菌有暴发性增殖的机会[27]。本研究中华按蚊B样本的细菌群落Chao1指数大于A样本支持此结论(表 2),2组样本间差异倍数 > 10的细菌门仅OP10,肠道优势菌属为不动杆菌属和泛菌属,B样本中Asaia属所占比例最高(40.85%),多项研究发现在伊蚊与按蚊的肠道中Asaia菌也普遍存在[7, 28-29]。本研究的中华按蚊B样本的细菌菌群多样性指数较小,可能是由于A样本为肉眼判断确定,是否曾经吸血不得而知,结果体现了现场样本的复杂之处。以往较多的研究是以在实验室养殖传代的蚊虫中分离鉴定细菌菌群,发现与现场采集的蚊虫在菌群结构上完全不同,前者在传代过程中会丢失更多[30],因此,现场的蚊虫肠道细菌菌群研究更有意义。
已有的研究发现蚊种和生态环境对肠道菌群群落结构影响不大。如本研究结果显示现场中华按蚊成蚊肠道中,变形菌门、厚壁菌门和拟杆菌门的种类为优势类群,与冈比亚按蚊雌蚊肠道[23]、实验室饲养斯氏按蚊(An. stephensi)、白端按蚊(An. albimanus)成蚊肠道的优势菌群一致[5, 31](变形菌门和拟杆菌门)。17个地点采集的冈比亚按蚊细菌菌群主要隶属变形菌门,丰度高的属是肠杆菌属Enterobacter和气单胞菌属Aeromonas,不同生态环境的菌群差异无统计学意义,仅丰富度和多样性指数有少许差别[32]。也有研究发现蚊虫肠道菌群多样性与生态环境、个体的摄食习惯、抗药性等存在一定相关性[7, 23]。检测发现现场成蚊肠道细菌菌群在蚊种及个体间的差异较大,而成蚊是如何获得的微生物,影响群落结构的因素等目前尚未明晰[33]。中华按蚊在我国分布广泛,部分群体间已出现明显的遗传分化[34]。本研究采自上海市嘉定区的中华按蚊成蚊肠道优势菌群是不动杆菌属、泛菌属和Asaia,而Gao等[17]研究报道的江苏、辽宁和云南省的中华按蚊成蚊“肠道核心菌群”是沙雷菌属,不同群体的菌群组成也存在差异,如:云南群体丰度最高的是Asaia(25.41%)和沙雷菌(16.21%),辽宁群体是沙雷菌(51.45%)和红球菌(Rhodococcus,4.34%),江苏群体是弓形菌(Arcobacter,77.03%)和沙雷菌(21.11%)[18]。上述不同群体与本研究的优势菌群存在差异的原因,可能与生态环境、取样方法相关,需要进一步积累数据解释和分析。
随着高通量测序技术的发展和应用,愈来愈多的蚊虫生活史阶段和不同生理期的共生菌群将得以揭示,蚊虫肠道菌群的生物学功能等方面的研究也取得了一些进展,如按蚊、共生菌群和疟原虫,三者之间的互作关系[4-5, 35]。但要深入理解蚊虫与共生微生物之间复杂的相互关系,仍面临以下挑战,即检测传病蚊虫共生的所有微生物组成情况[36],需要更好地描述非细菌微生物群落的特征,以及肠外微生物的功能等。
志谢 承蒙上海市嘉定区疾病预防控制中心徐友祥、海军军医大学朱钊民和伍桐等协助现场采集,特此志谢利益冲突 无
| [1] |
中国科学院中国动物志编辑委员会. 中国动物志.第八卷.昆虫纲. 双翅目: 蚊科(上)[M]. 北京: 科学出版社, 1997: 4-33. Editorial Board of Zoology of China, Chinese Academy of Sciences. Fauna sinica.Vol. 8. Insecta. Diptera: Culicidae 1[M]. Beijing: Science Press, 1997: 4-33. |
| [2] |
张丽, 丰俊, 夏志贵, 等. 2019年全国疟疾疫情特征分析及消除工作进展[J]. 中国寄生虫学与寄生虫病杂志, 2020, 38(2): 133-138. Zhang L, Feng J, Xia ZG, et al. Epidemiological characteristics of malaria and progress on its elimination in China in 2019[J]. Chin J Parasitol Parasit Dis, 2020, 38(2): 133-138. DOI:10.12140/j.issn.1000-7423.2020.02.001 |
| [3] |
曹俊, 刘耀宝, 曹园园, 等. 中国消除疟疾的持续挑战: 输入性疟疾[J]. 中国寄生虫学与寄生虫病杂志, 2018, 36(2): 93-96. Cao J, Liu YB, Cao YY, et al. Sustained challenge to malaria elimination in China: imported malaria[J]. Chin J Parasitol Parasit Dis, 2018, 36(2): 93-96. |
| [4] |
Gao H, Cui CL, Wang LL, et al. Mosquito microbiota and implications for disease control[J]. Trends Parasitol, 2020, 36(2): 98-111. DOI:10.1016/j.pt.2019.12.001 |
| [5] |
Romoli O, Gendrin M. The tripartite interactions between the mosquito, its microbiota and Plasmodium[J]. Parasit Vectors, 2018, 11: 200. DOI:10.1186/s13071-018-2784-x |
| [6] |
Sharma P, Rani J, Chauhan C, et al. Altered gut microbiota and immunity defines Plasmodium vivax survival in Anopheles stephensi[J]. Front Immunol, 2020, 11: 609. DOI:10.3389/fimmu.2020.00609 |
| [7] |
Boissière A, Tchioffo MT, Bachar D, et al. Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection[J]. PLoS Pathog, 2012, 8(5): e1002742. DOI:10.1371/journal.ppat.1002742 |
| [8] |
Dong YM, Manfredini F, Dimopoulos G. Implication of the mosquito midgut microbiota in the defense against malaria parasites[J]. PLoS Pathog, 2009, 5(5): e1000423. DOI:10.1371/journal.ppat.1000423 |
| [9] |
Bando H, Okado K, Guelbeogo WM, et al. Intra-specific diversity of Serratia marcescens in Anopheles mosquito midgut defines Plasmodium transmission capacity[J]. Sci Rep, 2013, 3: 1641. DOI:10.1038/srep01641 |
| [10] |
Gonzalez-Ceron L, Santillan F, Rodriguez MH, et al. Bacteria in midguts of field-collected Anopheles albimanus block Plasmodium vivax sporogonic development[J]. J Med Entomol, 2003, 40(3): 371-374. DOI:10.1603/0022-2585-40.3.371 |
| [11] |
Wang SB, Ghosh AK, Bongio N, et al. Fighting malaria with engineered symbiotic bacteria from vector mosquitoes[J]. Proc Natl Acad Sci USA, 2012, 109(31): 12734-12739. DOI:10.1073/pnas.1204158109/-/DCSupplemental |
| [12] |
Kajla MK. Symbiotic bacteria as potential agents for mosquito control[J]. Trends Parasitol, 2020, 36(1): 4-7. DOI:10.1016/j.pt.2019.07.003 |
| [13] |
李美, 汤林华. 中华按蚊实验室种群中肠内细菌菌群分析[J]. 中国寄生虫学与寄生虫病杂志, 2010, 28(2): 143-147. Li M, Tang LH. The midgut bacterial flora in lab-reared Anopheles sinensis[J]. Chin J Parasitol Parasit Dis, 2010, 28(2): 143-147. |
| [14] |
王丹丹. 中华按蚊肠道细菌分子多态性研究[D]. 武汉: 华中农业大学, 2008. Wang DD. Molecular diversity of the intestine bacterial community of Anopheles sinensis (Diptera: Culicidae)[D]. Wuhan: Huazhong Agricultural University, 2008. |
| [15] |
翟小战. 中华按蚊肠道细菌群落的研究[D]. 武汉: 华中农业大学, 2014. Zhai XZ. The intestinal bacterial flora community structure of Anopheles sinensis mosquitoes (Diptera: Culicidae)[D]. Wuhan: Huazhong Agricultural University, 2014. |
| [16] |
南春燕, 马雅军, 徐建农, 等. 中华按蚊幼虫肠道细菌宏基因组的组成研究[J]. 中国寄生虫学与寄生虫病杂志, 2013, 31(2): 114-119. Nan CY, Ma YJ, Xu JN, et al. Taxonomic composition of metagenomic community in the larval gut of mosquito Anopheles sinensis (Diptera: Culicidae)[J]. Chin J Parasitol Parasit Dis, 2013, 31(2): 114-119. |
| [17] |
Gao H, Bai L, Jiang YM, et al. A natural symbiotic bacterium drives mosquito refractoriness to Plasmodium infection via secretion of an antimalarial lipase[J]. Nat Microbiol, 2021, 6(6): 806-817. DOI:10.1038/s41564-021-00899-8 |
| [18] |
Bai L, Wang LL, Vega-Rodríguez J, et al. A gut symbiotic bacterium Serratia marcescens renders mosquito resistance to Plasmodium infection through activation of mosquito immune responses[J]. Front Microbiol, 2019, 10: 1580. DOI:10.3389/fmicb.2019.01580 |
| [19] |
Tran Q, Pham DT, Phan V. Using 16S rRNA gene as marker to detect unknown bacteria in microbial communities[J]. BMC Bioinformatics, 2017, 18(14): 499. DOI:10.1186/s12859-017-1901-8 |
| [20] |
Wang Y, Gilbreath Ⅲ TM, Kukutla P, et al. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya[J]. PLoS One, 2011, 6(9): e24767. DOI:10.1371/journal.pone.0024767 |
| [21] |
Champion CJ, Xu JN. The impact of metagenomic interplay on the mosquito redox homeostasis[J]. Free Radic Biol Med, 2017, 105: 79-85. DOI:10.1016/j.freeradbiomed.2016.11.031 |
| [22] |
Gimonneau G, Tchioffo MT, Abate L, et al. Composition of Anopheles coluzzii and An. gambiae microbiota from larval to adult stages[J]. Infect Genet Evol, 2014, 28: 715-724. DOI:10.1016/j.meegid.2014.09.029 |
| [23] |
Osei-Poku J, Mbogo CM, Palmer WJ, et al. Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya[J]. Mol Ecol, 2012, 21(20): 5138-5150. DOI:10.1111/j.1365-294X.2012.05759.x |
| [24] |
彭恒, 陈翰明, 陈辉莹, 等. 我国赫坎按蚊种团的分子鉴别及中华按蚊的区系分布研究[J]. 中国寄生虫学与寄生虫病杂志, 2020, 38(1): 58-66, 73. Peng H, Chen HM, Chen HY, et al. Molecular identification of Anopheles hyrcanus group and faunal distribution of An. sinensis (Diptera: Culicidae) in China[J]. Chin J Parasitol Parasit Dis, 2020, 38(1): 58-66, 73. DOI:10.12140/j.issn.1000-7423.2020.01.009 |
| [25] |
Wang Y, Qian PY. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies[J]. PLoS One, 2009, 4(10): e7401. DOI:10.1371/journal.pone.0007401 |
| [26] |
Li KL, Chen HY, Jiang JJ, et al. Diversity of bacteriome associated with Phlebotomus chinensis (Diptera: Psychodidae) sand flies in two wild populations from China[J]. Sci Rep, 2016, 6: 36406. DOI:10.1038/srep36406 |
| [27] |
de O Gaio A, Gusmão DS, Santos AV, et al. Contribution of midgut bacteria to blood digestion and egg production in Aedes aegypti (Diptera: Culicidae) (L.)[J]. Parasit Vectors, 2011, 4: 105. DOI:10.1186/1756-3305-4-105 |
| [28] |
Zouache K, Raharimalala FN, Raquin V, et al. . Bacterial diversity of field-caught mosquitoes, Aedes albopictus and Ae. aegypti, from different geographic regions of Madagascar[J]. FEMS Microbiol Ecol, 2011, 75(3): 377-389. DOI:10.1111/j.1574-6941.2010.01012.x |
| [29] |
Alfano N, Tagliapietra V, Rosso F, et al. Changes in microbiota across developmental stages of Aedes koreicus, an invasive mosquito vector in Europe: indications for microbiota-based control strategies[J]. Front Microbiol, 2019, 10: 2832. DOI:10.3389/fmicb.2019.02832 |
| [30] |
Akorli J, Namaali PA, Ametsi GW, et al. Generational conservation of composition and diversity of field-acquired midgut microbiota in Anopheles gambiae (sensu lato) during colonization in the laboratory[J]. Parasit Vectors, 2019, 12: 27. DOI:10.1186/s13071-019-3287-0 |
| [31] |
Kalappa DM, Subramani PA, Basavanna SK, et al. Influence of midgut microbiota in Anopheles stephensi on Plasmodium berghei infections[J]. Malar J, 2018, 17: 385. DOI:10.1186/s12936-018-2535-7 |
| [32] |
Zoure AA, Sare AR, Yameogo F, et al. Bacterial communities associated with the midgut microbiota of wild Anopheles gambiae complex in Burkina Faso[J]. Mol Biol Rep, 2020, 47: 211-224. DOI:10.1007/s11033-019-05121-x |
| [33] |
Coon KL, Vogel KJ, Brown MR, et al. Mosquitoes rely on their gut microbiota for development[J]. Mol Ecol, 2014, 23(11): 2727-2739. DOI:10.1111/mec.12771 |
| [34] |
Feng XY, Huang LB, Lin L, et al. Genetic diversity and population structure of the primary malaria vector Anopheles sinensis (Diptera: Culicidae) in China inferred by cox1 gene[J]. Parasit Vectors, 2017, 10: 75. DOI:10.1186/s13071-017-2013-z |
| [35] |
Gabrieli P, Caccia S, Varotto-Boccazzi I, et al. Mosquito trilogy: microbiota, immunity and pathogens, and their implications for the control of disease transmission[J]. Front Microbiol, 2021, 12: 630438. DOI:10.3389/fmicb.2021.630438 |
| [36] |
Mancini MV, Damiani C, Accoti A, et al. Estimating bacteria diversity in different organs of nine species of mosquito by next generation sequencing[J]. BMC Microbiol, 2018, 18: 126. DOI:10.1186/s12866-018-1266-9 |