 2021, Vol. 32
2021, Vol. 32扩展功能
文章信息
- 周秋明, 林琳, 董昊炜, 袁浩, 白洁, 李翔宇, 彭恒, 马雅军
- ZHOU Qiu-ming, LIN Lin, DONG Hao-wei, YUAN Hao, BAI Jie, LI Xiang-yu, PENG Heng, MA Ya-jun
- 基于多态微卫星DNA的中国雷氏按蚊群体遗传结构研究
- Genetic structure of Anopheles lesteri populations in China based on microsatellite loci
- 中国媒介生物学及控制杂志, 2021, 32(5): 526-532
- Chin J Vector Biol & Control, 2021, 32(5): 526-532
- 10.11853/j.issn.1003.8280.2021.05.004
-
文章历史
- 收稿日期: 2021-03-11




2 海军军医大学基础医学院病原生物学教研室, 上海 200433
2 Department of Medical Microbiology and Parasitology, College of Basic Medical, Naval Medical University, Shanghai 200433, China
雷氏按蚊(Anopheles lesteri)隶属于赫坎按蚊种团(An. hyrcanus group),由Ho等[1]于1962年在我国首次记录。赫坎按蚊种团存在许多形态难以鉴别的隐种,雷氏按蚊的分类地位也经历了多次修订[2-5]。雷氏按蚊嗜吸人血,在部分地区具有较高的媒介效能,是一种重要的传疟媒介[6]。《中国消除疟疾行动计划(2010-2020年)》提出,到2015年全国除云南省边境外实现无本地感染病例,到2020年全国实现消除疟疾的目标[7]。从2017年起,我国已经实现连续3年无本地感染病例报告[8]。2021年6月世界卫生组织确认中国已消除疟疾。在疟疾消除后,我国仍然面临着疟疾流行的风险,主要威胁来自于输入性病例由本地媒介传播造成的继发性感染[9]。因此,做好传疟媒介的监测和防制是巩固疟疾成果,防止疟疾再传播的关键环节之一。
雷氏按蚊在我国分布广,北至辽宁省,南至海南省都有分布,有研究显示不同分布地的雷氏按蚊生态习性和传病作用存在差异[6, 10]。因此,应用分子标志分析雷氏按蚊不同群体的遗传结构,有助于进一步阐释其差异产生的机制,也可预测其群体扩散迁徙的路线和趋势,对疟疾消除后媒介防控提供重要的理论基础。本课题组已应用随机扩增多态性DNA(random amplified polymorphic DNA,RAPD)、线粒体部分基因检测了我国雷氏按蚊群体的遗传变异,部分群体出现了显著的遗传分化,群体内遗传差异均大于群体间[11-13]。
微卫星DNA是广泛分布于基因组DNA中的短重复单元序列,是一种理想的解析蚊虫群体遗传结构的分子标志[14-21]。相对于RAPD和线粒体基因方法,微卫星DNA检测能提供更详细的遗传信息。本研究利用前期已分离的雷氏按蚊微卫星DNA,及筛选的多态性位点[22-23],对扩大收集的我国雷氏按蚊现场群体样本,检测群体内和群体间的遗传变异以及群体的稳定性和规模,分析群体的遗传结构,探讨其形成的影响因素,以期为我国疟疾消除后传疟媒介的监测和控制提供理论依据。
1 材料与方法 1.1 蚊虫采集与鉴定于1997-2010年间,多次在牲畜圈或人房,用吸蚊管人工吸取成蚊,采集信息见表 1和图 1。样本干燥后带回实验室,依据形态鉴定为赫坎按蚊种团成员种[6]。江苏盱眙雷氏按蚊样本为实验室品系,韩国江华岛雷氏按蚊样本由韩国Gi-Sik Min教授赠送。单只蚊成虫抽提基因组,依据核糖体DNA第2内转录间隔区(ribosomal DNA second internal transcribed spacer,rDNA-ITS2)特征进行分子鉴定[24],确定为雷氏按蚊的个体,进行后续微卫星检测。
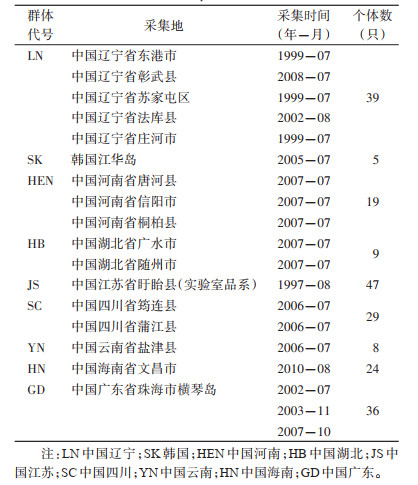
|

|
| 注:* 为雷氏按蚊的采集地;灰色部分为我国已知雷氏按蚊分布区域。LN辽宁;JS江苏;HEN河南;HB湖北;SC四川;YN云南;GD广东;HN海南。 图 1 本研究雷氏按蚊采集地示意图 Figure 1 Map of Anopheles lesteri collection sites |
| |
基因组提取试剂盒、TaqPCR预混液和DNA标记购自北京艾德莱生物科技有限公司,引物合成、序列测定和微卫星扩增由铂尚生物科技(上海)有限公司完成。
1.3 基因扫描对分子鉴定为雷氏按蚊的样本应用荧光标记PCR法,进行微卫星DNA的基因扫描,9个多态微卫星位点分别为:ANL5(EF620303.1)、ANL8(EF620306.1)、ANL10(EF620308.1)、ANL11(EF620309.1)、ANL12(EF620310.1)、ANL14(EF620312.1)、ANL16(EF620314.1)、ANL18(EF620316.1)和ANL20(EF620318.1),其扩增条件参照文献[22-23],Genemapper 4.0软件读取获得的扩增片段长度。
1.4 遗传差异分析记录和整理所有样本在每个微卫星位点的长度,应用Fstat软件计算等位基因数(A)和等位基因丰富度(Rs)、自交系数(FIS),期望杂合度(HE)和观察杂合度(HO),并统计每个微卫星位点的等位基因在群体中出现的频率。用Micro-Checker软件计算哑等位基因频率。使用Genepop软件分析每个群体中的每个位点是否符合哈代-温伯格平衡,以及成对位点的连锁不平衡情况。应用Genepop和Arlequin计算群体间的固定指数(FST),基因流(Nm)的计算公式: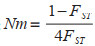
应用Structure软件包贝叶斯法推论群体的分支数(K),软件运行时选择混合选项,每个K值进行20次独立运算,参数设置如下:length of burnin period=10 000,number of MCMC reps after burnin=10 000,分支数的最大可能性(△K)用Harvester在线进行计算。
由于群体会存在近期的瓶颈和/或扩张的不稳定性,遗传差异计算时可能会产生明显的偏差,故突变漂移平衡(MDE)程度使用杂合子检验。应用Bottleneck软件,在MDE条件下,HE以10%~30%的重复单位两相模型(TPM)和逐步突变模型(SMM)来计算。基于杂合子过剩和连锁不平衡模型,应用NeEstimator软件计算长期有效群体规模(Ne)。
2 结果 2.1 雷氏按蚊群体本研究的雷氏按蚊采集地共17个,分子鉴定共得到雷氏按蚊样本216只,将地理距离相近的采集地样本合并,以9个群体进行后续分析(表 1),分别为韩国(SK),中国辽宁(LN)、河南(HEN)、湖北(HB)、江苏(JS)、四川(SC)、云南(YN)、海南(HN)和广东(GD)。
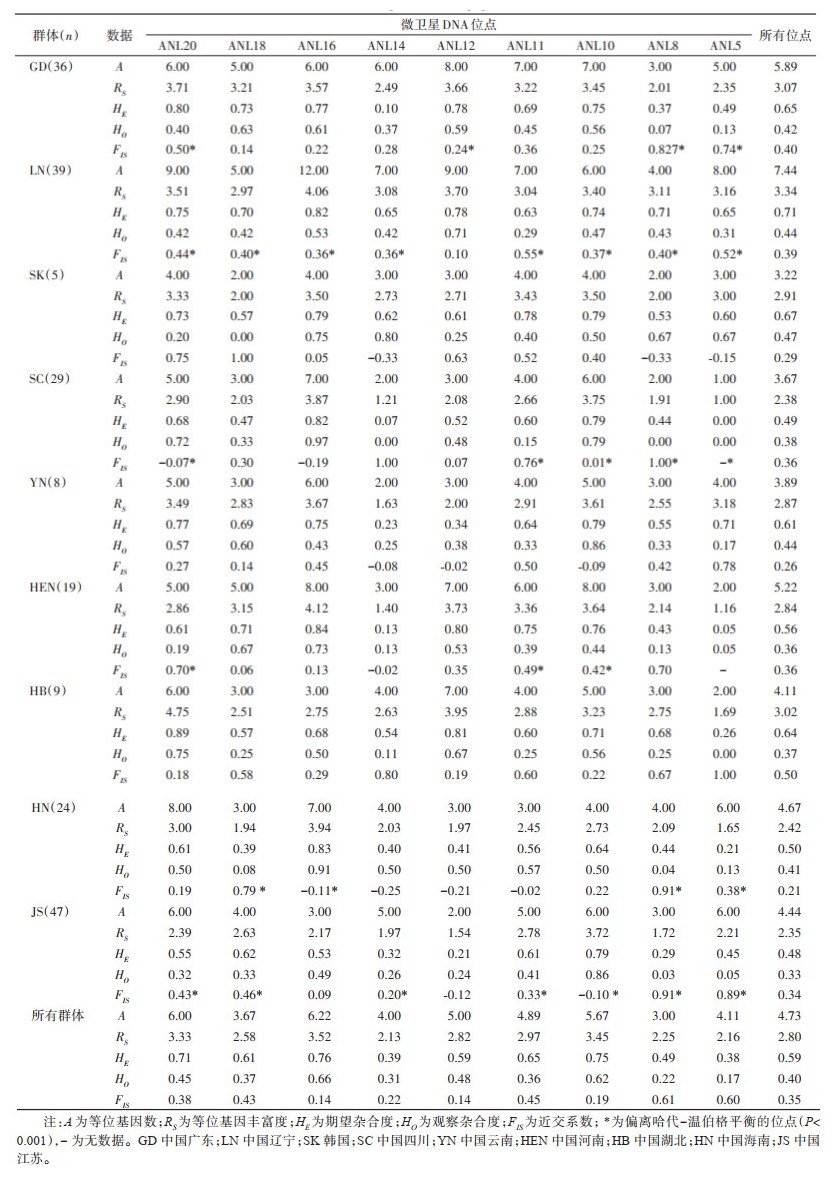
2.2 群体内遗传差异本研究检测的微卫星位点共9个,分别为ANL5、ANL8、ANL10、ANL11、ANL12、ANL14、ANL16、ANL18和ANL20。A和RS最大值分别为12.00和4.06(ANL16),最小值均为1.00(ANL5),平均值分别为4.73和2.80;平均等位基因数最少的群体是SK(3.22),最多的是LN(7.44)。雷氏按蚊各群体HE的范围为0.48(JS)~0.71(LN),平均值为0.59;HO的范围为0.33(JS)~0.47(SK),平均值为0.40;FIS范围为0.21(HN)~0.50(HB),平均为0.35。见表 2。
|
分析微卫星位点等位基因在各群体内出现的频率和自有等位基因的数目,结果显示位点ANL18和ANL14的所有等位基因在群体间共享,LN群体中自有等位基因最多,为9个,相反,YN群体无自有等位基因;哑等位基因频率最高的是HB群体ANL14位点(0.37),LN群体在9个位点均有哑等位基因。Fisher’s检验结果显示,ANL10与ANL11在所有群体中出现连锁关联,其余7个位点未检测到存在固定的连锁关系。
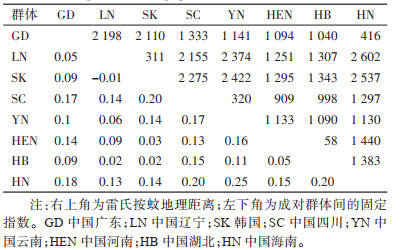
2.3 群体间遗传差异分析群体间遗传差异时,将实验室品系JS群体去除,得到的成对群体间FST范围为-0.01(LN-SK)~0.25(YN-HN)(表 3)。Nm数据显示,除了SC群体与HN、HB和YN群体间的数值< 1.00外,其余均 > 1.00,最大的数值出现在SK与LN群体之间。
|
Mantel检验结果显示,群体间的遗传差异与地理距离呈正相关关系(R2=0.002 9,P=0.360),提示群体遗传结构符合地理隔离模型(isolation-by-distance model)。分子变异等级分析结果显示,雷氏按蚊的遗传变异,群体内个体间的贡献为86.55%,远大于群体间(13.45%),FST值为0.13,Nm水平为1.67。
2.4 群体遗传结构Stucture分析的前提是假设所有微卫星位点无连锁关联,由于本研究ANL10与ANL11位点存在固定的连锁关系,故分别去除位点ANL10或ANL11后进行分析,结果显示△K值分别为4和2。若以0.8为阈值统计个体的归属情况,则低于0.8视为无法归类。△K=4时,无法归类的个体占32.54%,△K=2时,无法归类者占5.92%。结合群体内遗传差异的数据综合分析,本研究雷氏按蚊群体分为2个基因库较为合理(图 2),若以群体中个体归属基因库的比例分支,则第Ⅰ支包括LN和GD群体的部分个体(> 0.80),HEN、HB和SK的个别个体;第Ⅱ支包括SK、SC、YN、HEN、HB和HN群体的个体(> 0.80),以及LN和GD群体的个别个体。

|
| 注:色彩代表不同的分支;竖线代表个体;X轴代表群体;Y轴代表个体被分配至每个分支的概率;GD中国广东;LN中国辽宁;SK韩国;SC中国四川;YN中国云南;HEN中国河南;HB中国湖北;HN中国海南。 图 2 Structure贝叶斯分支分析结果(△K=2) Figure 2 Bayesian cluster analysis using Structure (△K=2) |
| |
基于微卫星突变SMM和TPM 2种模型的杂合子检验,结果显示HEN、HB、HN和LN群体经历了近期扩张。基于杂合子过剩模型计算群体规模,8个群体规模均为无穷大;而基于连锁不平衡模型,群体规模的范围为1.20(HB)~33.70(GD),总体为44.60,95%可信区间为40.60~49.00。
3 讨论雷氏按蚊在我国分布广,其范围是100°E以东,19°~42°N的18个省(直辖市、自治区)[6, 25]。本研究收集了我国东北、西南、华南和长江淮河流域等地的雷氏按蚊现场群体,尽量涵盖了雷氏按蚊的分布范围,与以往的研究报告相比[11-13],增加了海南岛的现场样本,更具代表性。多年的研究证明,rDNA-ITS2片段的序列差异具有理想的赫坎按蚊种团成员隐种鉴别解析度[24, 26-27],故基于rDNA-ITS2分子标志可以准确鉴别雷氏按蚊。
本研究应用9个微卫星DNA位点分析雷氏按蚊的群体遗传差异,平均等位基因数为4.73,群体的HE和HO最低的均为江苏群体,该群体为实验室品系,杂合度低,与长期驯化导致遗传背景趋于一致的现状相符,提示本研究应用的微卫星DNA位点具有合适的多态性。
本研究中,雷氏按蚊群体的平均近交系数为0.35,提示群体内更易发生近交,主要与雷氏按蚊呈点状分布,孳生地要求严格,较难发生远交有关。其中海南群体中的FIS值较低,表明该群体内个体间的远交机会更多,其采集地为海边,推测蚊虫是由渔船从异地携带而来,且经历了多次的迁入,定植后群体逐渐扩大。湖北、四川、河南、广东和辽宁群体FIS值较高,表明这些群体内近交的可能性较大,应是群体数量小导致;上述多个地区在20世纪后期均为疟疾的稳定流行区,且雷氏按蚊被认为是当地最重要的传播媒介[6],曾持续大量使用化学杀虫剂,使按蚊群体数量极度下降,这也可能是导致群体杂合子不足的重要因素。
比较不同分子标志检测的雷氏按蚊的群体遗传差异[11-13],虽然计算的相关指标大小不同,但趋势相同,均表明我国雷氏按蚊群体遗传变异水平中等;群体间遗传差异大小与地理距离呈正相关,群体结构符合地理隔离模型,群体结构形成的因素主要与分布地和生态习性有关。我国云南省地理地貌复杂,是著名的生物多样性的中心[28-29],本研究结果显示雷氏按蚊云南群体无自有等位基因,符合祖先群体的特征,若将云南群体的个体(第Ⅱ支)所属基因库设为原始基因库(图 2绿色),那么推测雷氏按蚊的迁移是从云南省向北、向东方向的路线,为适应当地气候和生态环境,在迁移中的群体出现一定程度的遗传变异,逐渐形成了不同的基因库(图 2红色)。与云南省地理距离越远的群体,隶属于不同基因库的个体比例越高,北到辽宁群体,南到广东群体时,几乎各占50.00%。海南群体则较为特殊,采集地为岛礁环境与其他群体存在地理屏障,且雷氏按蚊在海南岛的分布极为局限,检测到95.83%的个体属于原始群体的基因库,推测是由“建立者效应”(founder effect)所致,故海南群体与其他分布地的群体遗传差异较大。
不同蚊种生态习性不同,群体遗传和结构则完全不同,例如与雷氏按蚊同域分布的中华按蚊(An. sinensis),其幼蚊孳生地类型广泛,成蚊嗜吸牛血,群体数量大;基于分子特征检测的中华按蚊群体间基因交流充分,群体间遗传差异小,存在不明显的遗传结构,群体间差异与地理距离无对应关系[14, 30]。而雷氏按蚊幼蚊孳生地较中华按蚊要求严格,通常是多水草、有遮阴、水质清凉而富有沙石的静水和缓流中,也包括稻田、缓流的灌溉沟、渗出水浅潭以及清凉小水潭等,成蚊除吸人血外,也吸牛血[7],群体间遗传差异相对较大。不同遗传结构的雷氏按蚊传疟能力的差异有待进一步研究。
综上所述,我国雷氏按蚊群体的遗传变异水平中等,主要存在于群体内个体间;群体遗传结构明显,符合地理隔离模型,可分为2个基因库,云南群体为原始基因库,向东、北方向逐渐迁移和扩散,出现另一基因库的个体。在我国疟疾消除后阶段,雷氏按蚊群体仍需要密切监测和研究。
志谢 承蒙辽宁省东港市疾病预防控制中心(CDC)陈哲,广东省珠海市CDC黄利群,江苏省寄生虫病防治所周华云,海南省CDC曾林海,四川省CDC雷心田,云南省寄生虫病防治所顾云安,湖北省CDC黄光全和陈国英,河南省CDC李蓬,海军军医大学杨曼尼、吴静、樊勇等协助采集标本,一并志谢利益冲突 无
| [1] |
Ho C, Chou TC, Chen TH, et al. The Anopheles hyrcanus group and its relation to malaria in east China[J]. Chin Med J, 1962, 81: 71-78. |
| [2] |
许锦江, 冯兰洲. 我国赫坎按蚊类群的研究[J]. 昆虫学报, 1975, 18(1): 77-98. Xu JJ, Feng LZ. Study on the Anopheles hyrcanus group of mosquitoes in China[J]. Acta Entamol Sin, 1975, 18(1): 77-98. DOI:10.16380/j.kcxb.1975.01.013 |
| [3] |
Ma YJ, Xu JN. The Hyrcanus group of Anopheles (Anopheles) in China (Diptera: Culicidae): species discrimination and phylogenetic relationships inferred by ribosomal DNA internal transcribed spacer 2 sequences[J]. J Med Entomol, 2005, 42(4): 610-619. DOI:10.1093/jmedent/42.4.610 |
| [4] |
Hwang UW, Tang LH, Kobayashi M, et al. Molecular evidence supports that Anopheles anthropophagus from China and An. lesteri from Japan are the same species[J]. J Am Mosq Control Assoc, 2006, 22(2): 324-326. DOI:10.2987/8756-971X(2006)22[324:MESTAA]2.0.CO;2 |
| [5] |
Rueda LM, Wilkerson RV, Li CH. Anopheles (Anopheles) lesteri Biases and Hu (Diptera: Culicidae): neotype designation and description[J]. Proc Entomol Soc Wash, 2005, 107(3): 604-622. |
| [6] |
中国科学院中国动物志编辑委员会. 中国动物志. 第九卷. 昆虫纲. 双翅目: 蚊科(下)[M]. 北京: 科学出版社, 1997: 12-15, 31-34. Editorial Board of Zoology of China. Chinese Academy of Sciences. Fauna sinica. vol. 9. Insecta, Diptera: Culicidae Ⅱ[M]. Beijing: Science Press, 1997: 12-15, 31-34. |
| [7] |
中华人民共和国卫生部, 发展改革委, 教育部, 等. 中国消除疟疾行动计划(2010-2020年)[EB/OL]. (2010-05-26)[2021-03-10]. http://www.gov.cn/zwgk/2010-05/26/content_1614176.htm. Ministry of Health of the People's Republic of China, Development and Reform Commission, Ministry of Education, et al. Action plan for malaria eradication in China (2010-2020 years)[EB/OL]. (2010-05-26)[2021-03-10]. http://www.gov.cn/zwgk/2010-05/26/content_1614176.htm. |
| [8] |
张丽, 丰俊, 夏志贵, 等. 2019年全国疟疾疫情特征分析及消除工作进展[J]. 中国寄生虫学与寄生虫病杂志, 2020, 38(2): 133-138. Zhang L, Feng J, Xia ZG, et al. Epidemiological characteristics of malaria and progress on its elimination in China in 2019[J]. Chin J Parasitol Parasit Dis, 2020, 38(2): 133-138. DOI:10.12140/j.issn.1000-7423.2020.02.001 |
| [9] |
曹俊, 刘耀宝, 曹园园, 等. 中国消除疟疾的持续挑战: 输入性疟疾[J]. 中国寄生虫学与寄生虫病杂志, 2018, 36(2): 93-96. Cao J, Liu YB, Cao YY, et al. Sustained challenge to malaria elimination in China: imported malaria[J]. Chin J Parasitol Parasit Dis, 2018, 36(2): 93-96. |
| [10] |
卢钟山, 田桢干, 许庆华. 嗜人按蚊不同地理株基因组RAPD分析[J]. 口岸卫生控制, 2001, 6(1): 17-21. Lu ZS, Tian ZG, Xu QH. The genome RAPD analysis of different geographical strains of Anopheles anthropophagus[J]. Port Health Control, 2001, 6(1): 17-21. DOI:10.3969/j.issn.1008-5777.2001.01.012 |
| [11] |
杨曼尼, 马雅军. 基于mtDNA-COⅠ基因序列的雷氏按蚊分子群体遗传结构研究[J]. 昆虫学报, 2009, 52(9): 1000-1007. Yang MN, Ma YJ. Molecular population genetic structure of Anopheles lesteri (Diptera: Culicidae) based on mtDNA-COⅠ gene sequences[J]. Acta Entomol Sin, 2009, 52(9): 1000-1007. DOI:10.3321/j.issn:0454-6296.2009.09.009 |
| [12] |
马雅军, 宋关鸿, 李翔宇. 我国辽宁省嗜人按蚊群体与其他分布地群体的遗传分化研究[J]. 中国寄生虫病防治杂志, 2002, 15(6): 321-324. Ma YJ, Song GH, Li XY. Study on population genetic divergence of Anopheles anthropophagus between Liaoning and other distributions in China[J]. Chin J Parasit Dis Con, 2002, 15(6): 321-324. |
| [13] |
Yang MN, Ma YJ, Wu J. Mitochondrial genetic differentiation across populations of the malaria vector Anopheles lesteri from China (Diptera: Culicidae)[J]. Malar J, 2011, 10: 216. DOI:10.1186/1475-2875-10-216 |
| [14] |
Ma YJ, Yang MN, Fan Y, et al. Population structure of the malaria vector Anopheles sinensis (Diptera: Culicidae) in China: two gene pools inferred by microsatellites[J]. PLoS One, 2011, 6(7): e22219. DOI:10.1371/journal.pone.0022219 |
| [15] |
Ogola EO, Odero JO, Mwangangi JM, et al. Population genetics of Anopheles funestus, the African malaria vector, Kenya[J]. Parasit Vectors, 2019, 12(1): 15. DOI:10.1186/S13071-018-3252-3 |
| [16] |
Gao J, Zhang HD, Guo XX, et al. Dispersal patterns and population genetic structure of Aedes albopictus (Diptera: Culicidae) in three different climatic regions of China[J]. Parasit Vectors, 2021, 14(1): 12. DOI:10.1186/s13071-020-04521-4 |
| [17] |
Wei Y, Wang JT, Song ZY, et al. Patterns of spatial genetic structures in Aedes albopictus (Diptera: Culicidae) populations in China[J]. Parasit Vectors, 2019, 12(1): 552. DOI:10.1186/s13071-019-3801-4 |
| [18] |
Altamiranda-Saavedra M, Conn JE, Correa MM. Genetic structure and phenotypic variation of Anopheles darlingi in northwest Colombia[J]. Infect Genet Evol, 2017, 56: 143-151. DOI:10.1016/j.meegid.2017.11.011 |
| [19] |
Kaddumukasa MA, Wright J, Muleba M, et al. Genetic differentiation and population structure of Anopheles funestus from Uganda and the southern African countries of Malawi, Mozambique, Zambia, and Zimbabwe[J]. Parasit Vectors, 2020, 13(1): 87. DOI:10.1186/s13071-020-3962-1 |
| [20] |
Ambrose L, Hanson JO, Riginos C, et al. Population genetics of Anopheles koliensis through Papua New Guinea: New cryptic species and landscape topography effects on genetic connectivity[J]. Ecol Evol, 2019, 9(23): 13375-13388. DOI:10.1002/ece3.5792 |
| [21] |
Lühken R, Heitmann A, Jansen S, et al. Microsatellite typing of Aedes albopictus (Diptera: Culicidae) populations from Germany suggests regular introductions[J]. Infect Genet Evol, 2020, 81: 104237. DOI:10.1016/j.meegid.2020.104237 |
| [22] |
马雅军, 樊勇, 吴静. 雷氏按蚊多态微卫星DNA位点的筛选和特征[J]. 寄生虫与医学昆虫学报, 2008, 15(3): 150-153. Ma YJ, Fan Y, Wu J. Isolation and characterization of polymorphic microsatellite markers of Anopheles lesteri (Diptera: Culicidae)[J]. Acta Parasitol Med Entomol Sin, 2008, 15(3): 150-153. DOI:10.3969/j.issn.1005-0507.2008.03.005 |
| [23] |
Molecular Ecology Resources Primer Development Consortium, An J, Bechet A, et al. Permanent genetic resources added to molecular ecology resources database 1 October 2009-30 November 2009[J]. Mol Ecol Resour, 2010, 10(2): 404-408. DOI:10.1111/j.1755-0998.2009.02827.x |
| [24] |
彭恒, 陈翰明, 陈辉莹, 等. 我国赫坎按蚊种团的分子鉴别及中华按蚊的区系分布研究[J]. 中国寄生虫学与寄生虫病杂志, 2020, 38(1): 58-66, 73. Peng H, Chen HM, Chen HY, et al. Molecular identification of Anopheles hyrcanus group and faunal distribution of An. sinensis (Diptera: Culicidae) in China[J]. Chin J Parasitol Parasit Dis, 2020, 38(1): 58-66, 73. DOI:10.12140/j.issn.1000-7423.2020.01.009 |
| [25] |
马雅军, 徐建农. 中国按蚊的分类研究进展[J]. 中国媒介生物学及控制杂志, 2015, 26(5): 433-438. Ma YJ, Xu JN. Progress of taxonomic study on the anopheline mosquitoes in China[J]. Chin J Vector Biol Control, 2015, 26(5): 433-438. DOI:10.11853/j.issn.1003.4692.2015.05.001 |
| [26] |
伍桐, 马雅军. rDNA-ITS2序列作为中国按蚊分子鉴别特征的评述: Ⅰ按蚊亚属[J]. 寄生虫与医学昆虫学报, 2013, 20(1): 16-24. Wu T, Ma YJ. The second internal transcribed spacer of nuclear ribosomal DNA as a molecular identity for Chinese anopheline taxonomy: Ⅰsubgenus Anopheles species[J]. Acta Parasitol Med Entomol Sin, 2013, 20(1): 16-24. DOI:10.3969/j.issn.1005-0507.2013.01.004 |
| [27] |
马雅军, 杨频, 徐建农, 等. 应用形态、染色体和分子特征研究我国雷氏按蚊的分类鉴别(双翅目: 蚊科)[J]. 昆虫分类学报, 2005, 27(3): 199-208. Ma YJ, Yang P, Xu JN, et al. Identification of Anopheles lesteri in China (Diptera: Culicidae): morphologic characters, chromosome karyotype and molecular markers[J]. Entomotaxonomia, 2005, 27(3): 199-208. DOI:10.3969/j.issn.1000-7482.2005.03.007 |
| [28] |
董学书, 周红宁, 龚正达. 云南蚊类志(上卷)[M]. 昆明: 云南科技出版社, 2010: 42-48. Dong XS, Zhou HN, Gong ZD. The mosquito fauna of Yunnan China (Volume One)[M]. Kunming: Yunnan Science & Technology Press, 2010: 42-48. |
| [29] |
Feng XY, Huang LB, Lin L, et al. Genetic diversity and population structure of the primary malaria vector Anopheles sinensis (Diptera: Culicidae) in China inferred by cox1 gene[J]. Parasit Vectors, 2017, 10(1): 75. DOI:10.1186/s13071-017-2013-z |
| [30] |
常雪莲, 钟代斌, 李小聪, 等. 基于mtDNA-COⅠ基因序列分析我国中华按蚊种群的遗传结构[J]. 南方医科大学学报, 2015, 35(2): 234-238, 247. Chang XL, Zhong DB, Li XC, et al. Analysis of population genetic structure of Anopheles sinensis based on mitochondrial DNA cytochrome oxidase subunitⅠgene fragment[J]. J Southern Med Univ, 2015, 35(2): 234-238, 247. |