 2021, Vol. 32
2021, Vol. 32扩展功能
文章信息
- 杨小娜, 张琳, 侯学霞, 郝琴
- YANG Xiao-na, ZHANG Lin, HOU Xue-xia, HAO Qin
- 16S rDNA全长高通量测序在蜱媒病原生物多样性研究中的应用
- Application of 16S rDNA full-length high-throughput sequencing in the study of tick-borne pathogen biodiversity
- 中国媒介生物学及控制杂志, 2021, 32(4): 404-411
- Chin J Vector Biol & Control, 2021, 32(4): 404-411
- 10.11853/j.issn.1003.8280.2021.04.004
-
文章历史
- 收稿日期: 2021-03-23




蜱分布广泛,宿主繁多,可携带多种病原体,是传播病毒、螺旋体、立克次体等多种病原体的重要媒介,对人兽造成严重危害[1],是目前携带病原体种类最多的媒介生物,也是除蚊虫外影响最广泛的媒介生物[2]。然而,许多蜱媒病原体仍未发现和确认,对已知蜱媒病原体的流行状况、多样性和毒力也知之甚少[3]。由于大多数蜱媒病原体很难在实验室环境中培养,蜱本身很难根据形态学进行精确识别,我们对蜱病原体种群结构及基因组流行病学的理解仍然不全面,故有必要对蜱媒病原体进行微生物监测以评估人类和动物感染风险。16S核糖体DNA(16S rDNA)是细菌分类学研究中最常用的“分子钟”,其序列包含9个可变区和10个保守区。可变区因细菌而异,且变异程度与细菌的系统发育密切相关。通过检测16S rDNA的序列变异和丰度,可以了解环境样品中微生物群落多样性信息。基于16S rDNA的分析方法在微生物分类鉴定、微生态研究等方面起到重要作用,在蜱的微生物监测中,常常通过PCR方法对特异性基因的扩增和一代测序结合来鉴定病原体的存在,但这种方法提供的关于目标微生物的信息有限,并且只能检测特定的病原体。随着DNA测序技术的发展,以高通量低成本为主要特征二代测序,广泛应用于不能分类培养群落的菌种鉴定,是目前常用的16S rDNA测序平台(Illumina HiSeq/MiSeq),由于读长的限制,二代测序只能对16S rDNA的某一或某几个可变区进行测序,如V4/V3-V4/V1-V3/V4-V5等,在物种分类鉴定准确度上难以达到一代测序的水平[4]。以单分子和长读长为主要特征的三代测序可以实现高通量全长16S rDNA测序,不需要分离培养,即可获得群落中所有微生物的全长16S rDNA信息,达到一代测序的物种鉴定准确性和二代测序的应用广度。本研究利用三代测序技术PacBio全长测序平台获得数据,鉴定蜱媒病原体和内共生菌,并根据结果设计特异性病原体检测方法进行验证。
1 材料与方法 1.1 蜱采集与DNA提取2019年6月,在山西省祁县凉平寨采用布旗法捕获蜱约500只,根据蜱的形态学特征初步确定蜱种。随机选择50只蜱经75%乙醇溶液和无菌水洗涤后,破碎,利用DNeasy Blood & Tissue Kit(QIAGEN,GERMANY)分别提取基因组DNA,扩增蜱线粒体16S rDNA片段,精确鉴定蜱种。
1.2 16S rDNA全长测序与分析测序采用PacBio平台、高通量全长16S rDNA测序方法。将原始测序数据进行处理,得到较准确的一致性序列;利用barcode信息将来自不同样品的扩增子进行拆分,去除低质量的序列,得到高质量的扩增子序列(Tags)。将Tags序列比对到16S rDNA数据库(Ribosomal Database Project,RDP),去除嵌合体、重复序列,将相似度在97%以上的Tags聚类为一个操作分类单元(operational taxonomic unit,OTU),并统计每个OTU的丰度。将OTU代表序列与注释数据库美国国立生物技术信息中心(NCBI,20170709)进行比对注释,得到的OTU用于后续分析。
做OTU累积曲线图判断样本量是否足够。进行物种组成分析得到各样本物种组成及比例,分别在门、纲、目、科、属、种水平对各样品作物种丰度柱状图。对单个样品中物种进行Alpha多样性分析,包括Observed species指数、Chao指数、Ace指数、Shannon指数、Simpson指数以及Good coverage指数。对样品间多样性分析进行Beta多样性分析,利用非加权组平均法(unweighted pair-group method with arithmetic mean,UPGMA)做聚类树,直观展示样品的物种组成及差异情况。统计学分析及图形化展示使用R软件,确定当地优势蜱种携带病原体的特征。
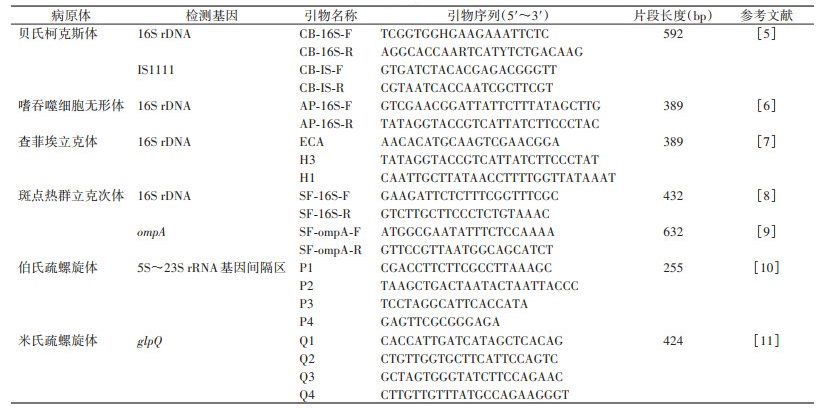
1.3 实验室验证根据16S rDNA全长测序分析结果,利用实验室检测方法对可能的蜱媒病原体进行鉴定。主要检测了柯克斯体属(Coxiella)中的贝氏柯克斯体(C. burnetii)、无形体属(Anaplasma)的嗜吞噬细胞无形体(A. phagocytophilum)、埃立克体属(Ehrlichia)的查菲埃立克体(E. chaffeensis)、立克次体属(Rickettsia)的斑点热群立克次体(spotted fever group rickettsia),以及疏螺旋体属的伯氏疏螺旋体(B. burgdorferi)和米氏疏螺旋体(B. miyamotoi)。扩增基因和引物见表 1。经PCR扩增后电泳,有目标条带者进行Sanger测序,将测序结果在NCBI的BLAST(https://blast.ncbi.nlm.nih.gov/Blast.cgi)中比对。
采集到的蜱经形态学鉴定均为血蜱。随机选取50只血蜱,通过扩增线粒体16S rDNA基因,经测序比对,确定其中47只为长角血蜱(Haemaphysalis longicornis),3只为日本血蜱(H. japonica)。
2.2 16S rDNA全长测序分析结果 2.2.1 蜱样本16S rDNA全长测序结果统计50只血蜱样本测序得到总序列数为481 195个,原始数据去除低质量的序列后,每个样品高质量的Tags均 > 1 000,平均为9 640条;Tags过滤并聚类为OTU后,50个样品中细菌总OTU数为943个。根据样品数目及OTU数目绘制物种累计曲线(图 1),随着样品数量的增加,能检测到的物种数量逐渐增加,最后曲线末端上升趋势趋于平缓,说明样本量足够,能用于表征该地区血蜱携带病原细菌的群落结构特征。

|
| 注:OTU表示操作分类单元。 图 1 山西省祁县50只血蜱携带病原体的物种累计曲线 Figure 1 Species accumulation curve of pathogens carried by 50 ticks in Qi county, Shanxi province, China |
| |
Alpha多样性包括Observed species指数、Chao指数、Ace指数、Shannon指数、Simpson指数以及Good coverage指数等,反映了血蜱中寄生细菌的物种复杂度。Observed species指数、Chao指数和Ace指数反映样品中群落的丰富度,50只血蜱中各自携带病原体的数量差异性较大,目前分析中可观测到的物种数在9(SX07)~332(SX62)之间。Shannon指数以及Simpson指数反映群落的多样性,其中21只血蜱多样性较高(Simpson指数 > 0.80)。根据Good coverage,50只血蜱样品文库的覆盖率均 > 97.00%,样品中序列没有被测出来的概率低。
在门、纲、目、科、属、种水平对各样品作物种丰度柱状图(图 2)。在50只血蜱中,从门水平分析,至少携带17个门的微生物,其中变形菌门(Proteobacteria)在每只血蜱中均携带且含量最多,物种丰度在72.93%~99.96%。从纲水平分析,至少携带26个纲的微生物,每只血蜱均携带γ-变形杆菌(Gammaproteobacteria)、α-变形杆菌(Alphaproteobacteria)。除SX7外的49只血蜱中γ-变形杆菌物种丰度均 > 50.00%,SX7中α-变形杆菌物种丰度高达97.78%。从目水平分析,至少携带46个目的微生物,每只血蜱均含有军团菌目(Legionellales)和假单胞菌目(Pseudomonadales),24只血蜱携带立克次体目(Rickettsiales)。从科水平分析,至少含有70个科的微生物,每只血蜱均含有柯克斯体科(Coxiellaceae)和假单胞菌科(Pseudomonadaceae),19只血蜱携带立克次体科(Rickettsiaceae),20只蜱携带无形体科(Anaplasmataceae);其中38只蜱中柯克斯体科物种丰度 > 50.00%。从属水平分析,至少携带100个属的微生物,每只蜱均含有柯克斯体属和假单胞菌(Pseudomonas),其中38只蜱柯克斯体属物种丰度 > 50.00%,19只蜱携带有立克次体属,17只蜱携带有埃立克体属,8只携带有无形体属;其中38只蜱柯克斯体属物种丰度 > 50.00%,SX07中立克次体属物种丰度为97.72%。种水平分析,50只蜱中共包括至少101个种的微生物,每只蜱均含有贝氏柯克斯体,8只蜱携带嗜吞噬细胞无形体,未检测到立克次体属、埃立克体属的相应基因型。

|
| 图 2 50只血蜱携带病原体的物种丰度柱状图 Figure 2 Histogram of species abundance of tick-borne pathogens in 50 ticks |
| |
Beta多样性分析是用来比较多个样品在物种多样性方面存在的差异大小。考虑序列的丰富度及各物种在样本中的含量,利用Weighted Unifrac距离矩阵做UPGMA聚类分析,50只蜱所携带病原体形成2个分支(图 3)。SX07样本含有97.72%的立克次体属和极少量柯克斯体、假单胞菌,与其他49个样品相比物种组成差异大,形成单独的分支。

|
| 图 3 样品聚类分析结果 Figure 3 Cluster analysis of samples |
| |
贝氏柯克斯体16S rDNA片段检测显示47只蜱呈阳性,测序后经BLAST比对,所得序列皆与柯克斯体内共生体(C. endosymbiont)有97.00%~99.60%的一致性。6个样品斑点热群ompA基因检测阳性。4个样品伯氏疏螺旋体5S~23S rRNA基因间隔区阳性,16个样品米氏疏螺旋体glpQ基因阳性。其他病原体检测均为阴性。将实验室验证结果与16S rDNA全长测序结果结合,结果见表 2。
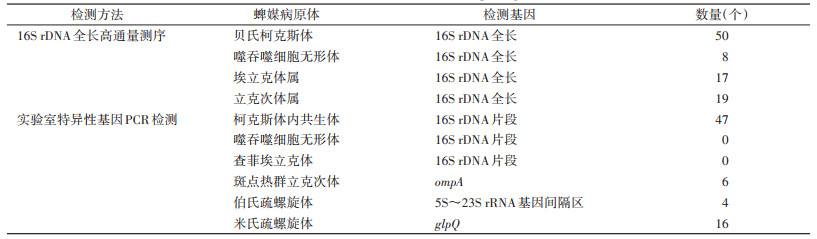
祁县位于山西省中部,有山脉、丘陵和平原等不同的地貌,宜林适牧、粮丰林茂,农业和畜牧业发达,生态环境适合血蜱的生存。血蜱是当地的优势蜱种,随机挑选50只血蜱进行物种鉴定、细菌16S rDNA全长测序分析,结果表明50只血蜱均携带贝氏柯克斯体、假单胞菌,8只血蜱携带嗜吞噬细胞无形体,有部分血蜱携带立克次体属、埃立克体,根据测序结果采用PCR方法验证,了解该地区优势蜱种携带微生物的多态性特点,为当地蜱媒病原体监测和防控提供基础数据。
16S rDNA全长测序分析表明50只血蜱均携带贝氏柯克斯体,通过PCR方法扩增16S rDNA基因片段有47只蜱检测阳性,经比对与柯克斯体内共生体同源性均 > 97.00%。柯克斯体与蜱保持共生关系,并可在蜱的所有生命阶段感染蜱,包括致病性贝氏柯克斯体和非致病性的柯克斯体内共生体[12]。贝氏柯克斯体是一种专性细胞内生长的革兰阴性菌,能在环境中长时间存活,是引起人类Q热的病原体[13]。它已从40多种硬蜱和至少14种软蜱中被分离出来,表明蜱在其传播中的重要性[14]。动物通过蜱叮咬感染贝氏柯克斯体,而人类主要通过接触污染的排泄物、粪便、直接接触分娩产物和空气而感染贝氏柯克斯体[15]。虽然尚未详细报告贝氏柯克斯体通过蜱叮咬直接传播给人类的情况[16],但贝氏柯克斯体已在受感染动物的牛奶、分娩产品、粪便和尿液中有过报告,人类可接触到这些动物,从而通过空气传播感染贝氏柯克斯体[13]。柯克斯体样内共生体已经在很多硬蜱和软蜱中发现,很多研究也发现其与蜱宿主生存和蜱媒病原体传播有密切关系[17]。本研究中50只血蜱均携带有贝氏柯克斯体,更有38只物种丰度 > 50.00%,可以推测该地宿主动物身上也携带此病原体,人类有感染的风险,这提示我们需要对宿主动物和当地居民进行贝氏柯克斯体感染的监测。
立克次体属,包括虱、鼠、蚤传播的斑疹伤寒群和蜱媒斑点热群[18],我们利用斑点热群的16S rDNA和外膜蛋白ompA基因进行鉴定,有6只蜱检测阳性,这6个样品在全长16S rDNA测序中均检测到携带立克次体属。埃立克体属,主要致病菌种为查菲埃立克体,能引起人单核细胞埃立克体病(Human monocytic ehrlichiosis,HME)[19];无形体属中主要致病菌为嗜吞噬细胞无形体,能引起人粒细胞无形体病(Human granulocytic anaplasmosis,HGA)[20]。经实验室16S rDNA片段监测,并未检出这2种病原体。
伯氏疏螺旋体是莱姆病的病原体,在世界范围内广泛存在,在我国至少有29个省(自治区、直辖市)发现伯氏疏螺旋体的感染,其中B. garinii、B. afzelii是我国的常见致病菌株[21]。米氏疏螺旋体是一种介于伯氏疏螺旋体和回归热螺旋体之间的一种特殊螺旋体[22],该病原体在我国研究并不多,只在东北牡丹江地区有报道[23],但其与伯氏疏螺旋体有共同的传播媒介和宿主,且能引起较为严重的疾病。本研究中在山西省祁县蜱中检测到2种疏螺旋体的存在,提示我们要加强该地区伯氏疏螺旋体、米氏疏螺旋体的监测,并了解当地宿主及人群的感染情况。
16S rDNA的高通量测序已成为微生物鉴定的标准方法,已被广泛用于研究蚊、蝇、蜱等媒介生物的微生物种群,研究病媒生物对病原体传播的影响[24-26]。这种方法能够鉴定低丰度和不可培养的细菌,能够在参考综合数据库的情况下提供高分辨率的检测。然而也有其局限性,容易产生微生物污染影响测序结果[27],数据量大,受限于参考数据库的准确性和全面性[26]。在本研究中使用的16S rDNA全长测序分析,除贝氏柯克斯体、嗜吞噬细胞无形体外,埃立克体属、立克次体属只匹配到属一级的分类信息,无法确定所携带的基因型,未检出疏螺旋体。根据测序结果设计实验室PCR检测方法,通过检测病原体特异性基因片段来判断病原体感染,具有良好的灵敏度和特异度。但只能检测特定的物种,效率较低,对DNA拷贝数较低者可能无法检出。在本研究中检测了常见的蜱媒病原体,并在50只蜱中检测到柯克斯体内共生体、伯氏疏螺旋体、米氏疏螺旋体、斑点热群立克次体的特异性基因片段。综合2种检测方法,分析对于蜱媒致病菌的检测,16S rDNA全长测序方法和病原体特异基因检测方法可互补,能更全面地了解蜱中病原体携带情况。
利益冲突 无
| [1] |
Mrzljak A, Novak R, Pandak N, et al. Emerging and neglected zoonoses in transplant population[J]. World J Transplant, 2020, 10(3): 47-63. DOI:10.5500/wjt.v10.i3.47 |
| [2] |
Rochlin I, Toledo A. Emerging tick-borne pathogens of public health importance: a mini-review[J]. J Med Microbiol, 2020, 69(6): 781-791. DOI:10.1099/jmm.0.001206 |
| [3] |
Ravi A, Ereqat S, Al-Jawabreh A, et al. Metagenomic profiling of ticks: Identification of novel rickettsial genomes and detection of tick-borne canine parvovirus[J]. PLoS Negl Trop Dis, 2019, 13(1): e0006805. DOI:10.1371/journal.pntd.0006805 |
| [4] |
Matsumoto T, Sugano M. 16S rRNA gene sequence analysis for bacterial identification in the clinical laboratory[J]. Rinsho Byori, 2013, 61(12): 1107-1115. |
| [5] |
Ni J, Lin HL, Xu XF, et al. Coxiella burnetii is widespread in ticks (Ixodidae) in the Xinjiang areas of China[J]. BMC Vet Res, 2020, 16(1): 317. DOI:10.1186/s12917-020-02538-6 |
| [6] |
Overzier E, Pfister K, Thiel C, et al. Anaplasma phagocytophilum in questing Ixodes ricinus ticks: comparison of prevalences and partial 16S rRNA gene variants in urban, pasture, and natural habitats[J]. Appl Environ Microbiol, 2013, 79(5): 1730-1734. DOI:10.1128/AEM.03300-12 |
| [7] |
Anderson BE, Sumner JW, Dawson JE, et al. Detection of the etiologic agent of human ehrlichiosis by polymerase chain reaction[J]. J Clin Microbiol, 1992, 30(4): 775-780. DOI:10.1128/JCM.30.4.775-780.1992 |
| [8] |
Yin XH, Guo SC, Ding CL, et al. Spotted fever group rickettsiae in Inner Mongolia, China, 2015-2016[J]. Emerg Infect Dis, 2018, 24(11): 2105-2107. DOI:10.3201/eid2411.162094 |
| [9] |
Roux V, Fournier PE, Raoult D. Differentiation of spotted fever group rickettsiae by sequencing and analysis of restriction fragment length polymorphism of PCR-amplified DNA of the gene encoding the protein rOmpA[J]. J Clin Microbiol, 1996, 34(9): 2058-2065. DOI:10.1128/JCM.34.9.2058-2065.1996 |
| [10] |
张琳, 苗广青, 侯学霞, 等. 巢式PCR和实时荧光定量PCR在莱姆病宿主动物监测中的应用评价[J]. 中国媒介生物学及控制杂志, 2018, 29(5): 425-427. Zhang L, Miao GQ, Hou XX, et al. Evaluation of nested PCR and real-time PCR in host surveillance of Lyme disease[J]. Chin J Vector Biol Control, 2018, 29(5): 425-427. DOI:10.11853/j.issn.1003.8280.2018.05.001 |
| [11] |
Schwan TG, Schrumpf ME, Hinnebusch BJ, et al. GlpQ: an antigen for serological discrimination between relapsing fever and Lyme borreliosis[J]. J Clin Microbiol, 1996, 34(10): 2483-2492. DOI:10.1128/JCM.34.10.2483-2492.1996 |
| [12] |
Klyachko O, Stein BD, Grindle N, et al. Localization and visualization of a coxiella-type symbiont within the lone star tick, Amblyomma americanum[J]. Appl Environ Microbiol, 2007, 73(20): 6584-6594. DOI:10.1128/AEM.00537-07 |
| [13] |
Maurin M, Raoult D. Q fever[J]. Clin Microbiol Rev, 1999, 12(4): 518-553. DOI:10.1128/CMR.12.4.518 |
| [14] |
Bola os-Rivero M, Carranza-Rodríguez C, Rodríguez NF, et al. Detection of Coxiella burnetii DNA in peridomestic and wild animals and ticks in an endemic region (Canary Islands, Spain)[J]. Vector Borne Zoonotic Dis, 2017, 17(9): 630-634. DOI:10.1089/vbz.2017.2120 |
| [15] |
Norlander L. Q fever epidemiology and pathogenesis[J]. Microbes Infect, 2000, 2(4): 417-424. DOI:10.1016/S1286-4579(00)00325-7 |
| [16] |
Pacheco RC, Echaide IE, Alves RN, et al. Coxiella burnetii in ticks, Argentina[J]. Emerg Infect Dis, 2013, 19(2): 344-346. DOI:10.3201/eid1902.120362 |
| [17] |
Zhong JM. Coxiella-like endosymbionts[J]. Adv Exp Med Biol, 2012, 984: 365-379. DOI:10.1007/978-94-007-4315-1_18 |
| [18] |
Roux V, Raoult D. Genotypic identification and phylogenetic analysis of the spotted fever group rickettsiae by pulsed-field gel electrophoresis[J]. J Bacteriol, 1993, 175(15): 4895-4904. DOI:10.1128/JB.175.15.4895-4904.1993 |
| [19] |
Ewing SA, Johnson EM, Kocan KM. Human infection with Ehrlichia canis[J]. N Engl J Med, 1987, 317(14): 899-900. DOI:10.1056/NEJM198710013171412 |
| [20] |
Dumler JS, Barbet AF, Bekker CPJ, et al. Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and 'HGE agent' as subjective synonyms of Ehrlichia phagocytophila[J]. Int J Syst Evol Microbiol, 2001, 51(6): 2145-2165. DOI:10.1099/00207713-51-6-2145 |
| [21] |
耿震, 万康林. 莱姆病流行病学研究新进展[J]. 中国自然医学杂志, 2017, 9(2): 158-160. Geng Z, Wan KL. New advances in epidemiological studies of Lyme disease[J]. Chin J Nat Med, 2017, 9(2): 158-160. |
| [22] |
Krause PJ, Fish D, Narasimhan S, et al. Borrelia miyamotoi infection in nature and in humans[J]. Clin Microbiol Infect, 2015, 21(7): 631-639. DOI:10.1016/j.cmi.2015.02.006 |
| [23] |
Jiang BG, Jia N, Jiang JF, et al. Borrelia miyamotoi infections in humans and ticks, northeastern China[J]. Emerg Infect Dis, 2018, 24(2): 236-241. |
| [24] |
Duguma D, Hall MW, Rugman-Jones P, et al. Developmental succession of the microbiome of Culex mosquitoes[J]. BMC Microbiol, 2015, 15: 140. DOI:10.1186/s12866-015-0475-8 |
| [25] |
Geiger A, Fardeau ML, Grebaut P, et al. First isolation of Enterobacter, Enterococcus, and Acinetobacter spp. as inhabitants of the tsetse fly (Glossina palpalis palpalis) midgut[J]. Infect Genet Evol, 2009, 9(6): 1364-1370. DOI:10.1016/j.meegid.2009.09.013 |
| [26] |
Couper L, Swei A. Tick microbiome characterization by next-generation 16S rRNA amplicon sequencing[J]. J Vis Exp, 2018(138): e58239. DOI:10.3791/58239 |
| [27] |
Salter SJ, Cox MJ, Turek EM, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses[J]. BMC Biol, 2014, 12: 87. DOI:10.1186/s12915-014-0087-z |