 2021, Vol. 32
2021, Vol. 32扩展功能
文章信息
- 栗冬梅, 周若冰, 李寿江, 鲁亮, 饶华祥, 宋秀平, 李庆多, 刘起勇
- LI Dong-mei, ZHOU Ruo-bing, LI Shou-jiang, LU Liang, RAO Hua-xiang, SONG Xiu-ping, LI Qing-duo, LIU Qi-yong
- 纳米孔测序实时检测鼠传巴尔通体
- Real-time detection of rodent-borne Bartonella by nanopore sequencing
- 中国媒介生物学及控制杂志, 2021, 32(4): 390-397
- Chin J Vector Biol & Control, 2021, 32(4): 390-397
- 10.11853/j.issn.1003.8280.2021.04.002
-
文章历史
- 收稿日期: 2021-03-02




2 青海省疾病预防控制中心, 青海 西宁 810007;
3 长治医学院, 山西 长治 046000
2 Qinghai Center for Disease Control and Prevention, Xining, Qinghai 810007, China;
3 Changzhi Medical College, Changzhi, Shanxi 046000, China
在传染病诊断、病原体检测与监测过程中,检测的时效性和准确性同等重要。特别是在常规监测、疫情暴发处理和追踪溯源时,快速的检测手段起着至关重要的作用。测序技术经历了一代、二代至三代的蓬勃发展后,病原宏基因组学概念应运而生,相关技术方法已经在病原体识别中有所应用[1-2]。二代测序(next-generation sequencing,NGS),是较早应用于病原体检测与鉴定的测序技术,目前比较成熟,以Illumina测序为主。该技术对设备安装和运行环境要求较高,读长较短,测序时间相对较长,测序文库制备和数据处理与分析较复杂,对专业技术要求较高[3-4]。近年来,三代纳米孔测序技术(nanopore sequencing)凭借其读长长、操作简便、实时读取测序数据和入门简单的特点脱颖而出,也在病原体检测方面展露出良好的前景[5]。特别是“掌上”纳米孔MinION测序仪具有小巧、操作便捷等优势,更加适用于现场工作。目前已实现在太空站、北极冰川这样的极端环境中成功测序[6]。
纳米孔测序的原理是在纳米孔两端施加一个外加电场,当不同碱基进入蛋白纳米孔时,引起电流不同程度变化,通过识别这种电流信号变化判断穿越纳米孔的碱基类别,从而确定整条核苷酸序列组成。Kilianski等[7]用MinION测序仪对痘病毒(Poxvirus)和大肠埃希菌(Escherichia coli)培养物进行扩增子测序,证明该技术可以准确地识别和区分病毒及细菌种类。近2年纳米孔测序技术在水体、动植物和其他环境样品宏基因组、病原体检测鉴定方面已经有越来越多的实际应用。例如,在非洲埃博拉[8]、拉沙热[9]和南美寨卡病毒病暴发疫情中快速甄别病原体[10],在畜牧业中对口蹄疫病毒进行分型研究以及在临床上对各种人体组织和体液进行检测以辅助诊断[11-12]。
测序技术用于临床感染性疾病病原体检测的优势在于无偏、快速地非靶向检测。但是有时鸟枪式宏基因组测序法对于一些低核酸丰度的组织或体液样品适用较差。针对这种情况,基于扩增子的宏基因组测序也许是一种更好的解决办法,特别是对于细菌的16S核糖体RNA(16S ribosomal RNA,16S rRNA)扩增子测序既可以降低背景基因组干扰又能够放大细菌存在的信号而提高检测效率[13-14]。由于16S扩增子纳米孔测序的种种优势和易操作性,本研究测试了MinION 16S扩增子测序是否适合直接从鼠类组织样品中快速检测细菌。我们选取巴尔通体(Bartonella spp.)培养阳性的样品,提取脾和肺组织DNA基因组,应用16S rRNA序列的通用引物对其进行常规PCR扩增和一代桑格测序(Sanger sequencing),同时进行纳米孔测序,评估16S扩增子纳米孔测序技术在鼠传病原体检测中的可行性。
1 材料与方法 1.1 样品来源选取2018年7月采集自青海省海西蒙古族藏族自治州(海西州)乌兰县、格尔木市的16只巴尔通体分离培养阳性的啮齿动物样品[15],其中14只为长尾仓鼠(Cricetulus longicaudatus)、2只为子午沙鼠(Meriones meridianus)。分别提取脾(P)、肺(F)组织核酸16份,组织在核酸提取前-80 ℃保存,未曾冻融。样品信息及巴尔通体菌株鉴定情况见表 1。

应用磁珠法提取动物脾、肺组织核酸(组织基因组DNA提取试剂盒AU19014,北京百泰克生物技术有限公司),应用Qubit 4荧光计(Invitrogen)和配套试剂盒(Qubit dsDNA HS Assay Kit)测定核酸浓度。
1.3 16S rRNA基因常规PCR扩增和测序应用引物fDl(5x-AGAGTTTGATCCTGGCTCAG-3x)-rDl(5x-AAG GAGGTGATCCAGCC-3x)[16]分别对脾和肺组织核酸扩增。反应体系50 μl,包括2×EasyTaqTM PCR SuperMix(北京全式金生物技术有限公司)25 μl、正反向引物各1 μl、基因组DNA 10 μl、无核酸酶水13 μl。PCR反应条件为95 ℃预变性1 min;95 ℃变性20 s,55 ℃退火30 s,65 ℃延伸2 min,25个循环;65 ℃延伸5 min(SensoQuest Labcycler PCR仪,德国圣欧)。1%的琼脂糖凝胶电泳100 V、30 min观察PCR产物,约1 500 bp扩增带阳性产物双向测序(北京奥科鼎盛生物科技有限公司)。应用BLASTn在线(www.ncbi.nlm.nih.gov)进行核酸序列同源性搜索比较。
1.4 Nanopore文库构建、测序和数据处理 1.4.1 文库构建Nanopore测序文库构建包括PCR扩增步骤,全部应用16S条码试剂盒(16S Barcoding Kit SQK-RAB204,牛津纳米孔技术公司Oxford Nanopore Technologies,ONT),使用含有条形码和5x末端标签的特异性16S引物(27F:5x-AGAGTTTGA TCMTGGCTCAG-3x[17]和1492R:5x-GGTTACCTTGTTACGACTT-3x[18]),对组织核酸DNA进行PCR扩增,实验操作参考操作手册(RAB_9053_v1_revC_19Dec2017),略作修改。PCR反应体系50 μl,包括LongAmp Taq 2×Master Mix(NEB)25 μl、基因组DNA 10 μl、16S barcode primer 1 μl和无核酸酶水14 μl。PCR反应条件同上述16S rRNA基因常规PCR扩增。取1 μl PCR产物定量浓度,将剩余产物全部转入1.5 ml低吸附离心管,加入30 μl磁珠(Agencourt AMPure XP Reagent,Beckman),室温混匀孵育后用70%乙醇溶液清洗,晾干残余乙醇后加入3~10 μl 10 mmol/L Tris-HCl(pH 8.0,含50 mmol/L NaCl)重悬磁珠,上清液即为纯化后的核酸。等体积吸取所有样品管中核酸,总体积15.5 μl,混匀后取1 μl定量浓度,依浓度值相应加入1.0~1.4 μl RAP,室温孵育5 min后加入34 μl Sequencing Buffer(SQB)、25.5 µl Loading Beads(LB),制备成总体积75 μl测序文库。
1.4.2 测序和数据处理按照操作手册将30 μl Flush Tether(FLT,Flow Cell Priming Kit,ONT)和1管Flush Buffer(FLB,Flow Cell Priming Kit,ONT)混匀,制备成测序缓冲液(priming mix),从priming port加入到测序芯片(Flow Cells R.9.4)中,然后将75 μl DNA文库从SpotON sample port加入到芯片中,关闭SpotON sample portpriming port,将芯片装载到MinION测序仪上进行测序。
测序软件为MinKNOW(MinION Release 19.06.7),应用Albacore软件实时识别碱基(Basecalling),运行14~20 h,起始电压180 mV,输出fast5和fastq文件。运行EPI2ME客户端(epi2 me-agent-v.2019.7.9-2549693),选择16S流程(FASTQ 16S v3.2.1)用于数据质控(Basecalling 1D-v2.2.8)和分类(16S Classification-v2.2.13),最小质量值(Min qscore)为7,选择检测barcode 16S RAB204,检测文件为*.fastq格式。进入EPI2ME网站(https://epi2me.nanoporetech.com)实时查看结果报告。
1.5 测序芯片flowcell清洗应用芯片清洗试剂盒(Washing kit,EXP-WSH002,ONT)按照操作说明对测序后的芯片进行清洗。应用溶液A(Solution A)清洗芯片后可以加入溶液B(Solution B)立即进行文库加载测序,也可以加入储存缓冲液(Storage Buffer)置于4~8 ℃保存备用。
2 结果 2.1 16S rRNA基因常规PCR扩增、测序及鉴定应用引物fDl-rDl分别扩增鼠脾和肺组织核酸,均出现约1 500 bp扩增带;该扩增带产物切胶纯化后测序,BLAST搜库(nt/nr)显示,在肺和脾组织样品中鉴定出巴尔通体的样品数量分别为11和9份,检出率分别为68.8%和56.2%;其中5份样品的脾和肺组织均鉴定出巴尔通体;4份肺和7份脾样品鉴定出根瘤菌和苍白杆菌等其他种属的细菌,有1份肺组织样品扩增产物测序无信号。见表 2。

全部样品核酸浓度在5.8~732.0 ng/μl,16S测序试剂盒含有12个测序样品标识(Barcode,BC01~12),每次最多操作12份样品。全部测序样品总数据量〔total yield(bases)〕为22.2~988 400 K,reads数量为38~647 576个,平均质量得分(average quality score)为6.4~9.9,数据产出主要在12 h以内(图 1),最早30 min即可在线(https://epi2me.nanoporetech.Com)观察到分类数据。序列长度集中在1 200~1 600 bp,以1 500 bp的reads为主,数量明显多于其他长度。见图 2。

|
| 注:数据结果由EPI2ME中QC and Barcoding[rev.3.10.2]分析而得。 图 1 测序数据产出的时间分布 Figure 1 Temporal distribution of sequencing data output |
| |

|
| 注:不同颜色代表不同批次测序。 图 2 16S扩增子Nanopore测序片段大小分布 Figure 2 Distribution of fragment size of 16S amplicon using nanopore sequencing |
| |
每份样品用于分类分析的reads数为4~609 424个不等,以巴尔通体检出为主,reads数为1~77 833个,其中脾组织平均为7 835个,肺组织平均为4 203个;巴尔通体鉴定的平均准确率(average accuracy)为79.2%~92.0%。见表 3。
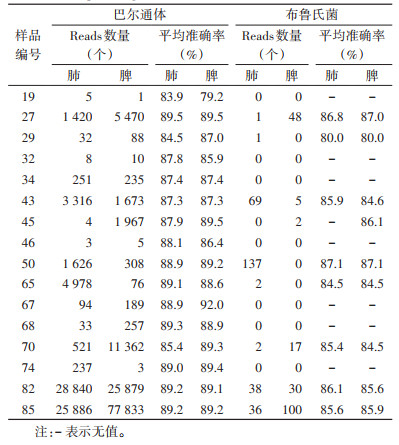
除巴尔通体外还鉴定出布鲁氏菌(Brucella sp.)和其他一些细菌。Reads数量较多的为支原体(Mycoplasma sp.)、苍白杆菌(Ochrobactrum sp.)、戴尔福特菌(Delftia sp.)、食酸菌(Acidovorax sp.)、假单胞菌(Pseudomonas sp.)和短波单胞菌(Brevundimonas sp.)等。见图 3。

|
| 注:分类数据结果由EPI2ME中16S Classification[rev.3.0.0]分析而得。 图 3 MinION纳米孔测序结果分类 Figure 3 Taxonomic tree based on nanopore MinION sequencing results |
| |
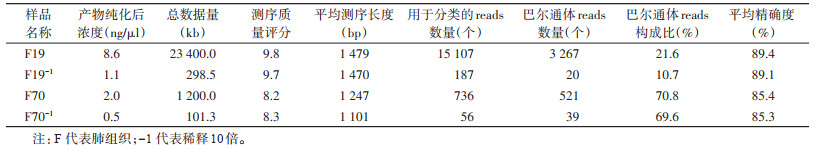
选择2个样品的核酸稀释10倍后扩增,纯化后用于制备测序文库的产物浓度较原始样品降低4~8倍。2个样品成功用于分析的reads差别较大,总数据量和reads数量明显降低1个数量级以上;样品核酸稀释前后所测序列平均长度基本一致;在稀释后的样品中用于分类的reads数明显减少1个数量级以上,鉴定精确度 > 85.0%(表 4)。样品稀释前后的测序质量和分类精确度值分别 > 8和 > 80.0%。
|
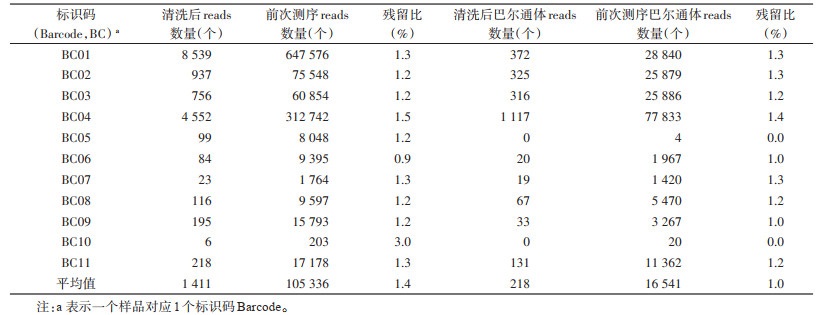
芯片清洗前后总reads的平均值分别为105 336和1 411个,残留比为0.9%~3.0%,平均值为1.4%;检出巴尔通体的reads平均值分别是16 541和218,残留比为0~1.4%,平均值为1.0%。见表 5。
|
啮齿动物是巴尔通体的主要宿主动物之一,目前从其体内分离到20余种巴尔通体,在鼠群中感染率相当高[19],有些鼠传巴尔通体具有致病性,如格拉汉姆巴尔通体[20-21]和伊丽莎白巴尔通体[22-23],是啮齿动物中常见的鼠传巴尔通体。我们选择巴尔通体作为指示菌检验纳米孔测序效果,较之于人工模拟样品能更好地反应样品检测的实际情况[24],是较为理想的自然样品。啮齿动物是很多致病菌的自然宿主,能传播包括鼠疫、钩端螺旋体病、恙虫病、地方性斑疹伤寒、野兔热和巴尔通体病在内的多种疾病[25],检测和监测相关病原体在鼠群中的流行动态是预防和控制这些传染病发生和流行的关键一环,高效的检测和监测手段是这一环节中应该具备的基本条件。
目前,对于很多病原体已具备较为高效的检测技术,如常规PCR和荧光定量PCR、多种等温扩增技术、多重检测技术、芯片和微流控技术[26],这些方法均是针对特异性核酸的靶标性检测方法,无法针对未知病原体进行筛检。二和三代高通量16S扩增子和鸟枪法宏基因组测序已广泛应用于环境样品的物种丰度和组成结构研究。近年关于病原体检测识别的报道和研究日渐增多,对于应用于临床诊断和实验室研究的宏基因组(病原)测序在国内外已形成了系统的操作标准并得到专家的广泛认同[27-28],逐渐呈现推广普及的趋势。因为三代测序发展较晚,所以目前应用于病原体检测的二代测序更多,但是三代测序仪MinION因测序片段长读长、方便携带和检测时间短而具有其独特的优势[29-31]。为评价该技术用于鼠传病原体检测的可行性,本研究选择了巴尔通体感染阳性的鼠类样品进行检测。
MinION测序后30 min内开始产生数据,同时进行碱基识别和分类分析。序列长度以1 500 bp的reads为主,数量明显多于其他长度,说明扩增、测序文库制备、上样和实时芯片测序步骤没有问题,产生了预期长度片段。全部脾和肺组织样品在纳米孔测序中均检测巴尔通体,不同样品的巴尔通体reads数差别比较大,平均准确率为79.2%~92.0%。此外,纳米孔测序不但检出巴尔通体,数个样品中脾和肺组织中还检测到布鲁氏菌,该菌是布鲁氏菌病的致病菌,动物宿主多为牛、羊、猪和犬,啮齿动物带菌虽不多见,但也有报道[32-33]。除巴尔通体和布鲁氏菌这2类致病菌外,还鉴定到一些环境微生物。宏基因组测序分析比对出大量菌群,包括很多环境微生物也是常见的现象,如何判断其与样品的关系问题,包括对一些不明原因的、新发病原体的确认,众多背景细菌会干扰对结果的判断,特别是与疾病的关联,要结合综合判断。应该结合样品的背景信息、临床、流行病学调查信息、病理学以及研究的目的。在宏基因组测序中大量数据的产生与测序技术和生物信息分析相关,但不是关键问题,对结果意义的判断主要取决于对研究目标的认知和相关知识库中的储备。
一般来说,待测样品带菌与否和带菌量多少直接影响最后的检测结果。16S扩增子测序的优势就在于可以放大检测样品全基因组中细菌的成分,易于检出和识别,即便如此,样品中靶核酸起始浓度也会影响测序结果。本研究中所用16S扩增子纳米孔测序16S条码试剂盒推荐起始核酸用量为10 ng,但是实际样品情况复杂,含有大量宿主动物的背景核酸,病原体核酸含量未知,如果盲目参照试剂盒操作手册稀释样品或许影响检出效果。本研究选择2个样品进行稀释,测试实验结果发现稀释原始样品浓度可降低测序数据量,特别是用于分类的reads明显减少,说明起始样品浓度是影响测序结果的直接因素,对序列长度分布和分类分析影响不大。样品经稀释后平均reads长度基本没有变化,不影响每个reads搜库分类鉴定,但会影响丰度较低的靶标,导致在样品中低丰度的病原体不能被检出,因此我们建议这类样品在第一步PCR扩增前不需要稀释。
测序芯片可以重复使用,这样可以增加芯片的使用效率和最大数据产出,ONT公司为此提供了芯片清洗方法和相应试剂盒,说明这一步骤可以去除大部分前次文库中的核酸片段。此外,在少量研究报道中也提到了关于测序芯片重复使用会产生残留的问题[34-35],也有研究者在ONT公司网站上的Community中讨论关于残留问题,但这方面的详细数据较少。在本研究中,我们提供了较为详细的数据,发现经过清洗的芯片再次应用时,可见上次测序实验产生的少量reads,在分析过程中可通过Barcode将其排除,但是如果2次实验均使用同样的Barcode,则无法鉴别Barcode相同的样品而产生数据混淆。这种情况出现时,如果用于分析样品细菌群落结构,对于丰度较高的细菌物种影响不大,但低丰度物种则可产生明显干扰。如果用于样品中病原体的检测则可能会出现假阳性结果,直接导致错误判断。
在测序过程中发现,上样操作非常重要,如果将气泡引入测序芯片,导致纳米孔堵塞,测序“活孔(active pores)”数量显著减少,测序质量明显下降。MinION测序成功与否的一个关键决定因素是在测序启动后执行MUX(Multiplexer)扫描时识别的有效纳米孔数目[6]。每个测序芯片包含2 048个纳米孔,由于生产、储存、运输和操作等因素会导致实际“活孔”数量减少,因此,在使用前需要进行质检,观察“活孔”数量,低于800个,为不合格芯片。我们在使用过程中没有遇到过 > 2 000个“活孔”的芯片,通常在1 000~2 000个范围内,是可以正常测序的。
16S rRNA,是原核生物核糖体中30S亚基的组成部分,因其结构与功能的高度保守性,可反映物种间的亲缘关系,常被用作细菌系统分类研究中的分子钟,有“细菌化石”之称。16S扩增子测序相较鸟枪法宏基因组测序操作简单、快速、数据量少和分析方便,既可以规避对仪器设备和操作人员的高要求,又可以一次性扩大病原体检测范围。本研究应用小量样本尝试评估此技术在鼠传细菌性病原检测中的应用,初步认为具有可行性。下一步,有必要进行更多大样本感染样品的宏基因组测序,加快对这一新检测手段进行客观评判,标准化地应用于传染病病原体检测和监测工作中。MinION的便携式测序使得临床的即时检验(point-of-care testing,POCT)、野外现场样品实时微生物检测以及环境样品微生物组成的实时监控等需求成为现实。当然,在实验过程中试剂冷藏、网络畅通和电力供应充足等需求必须得到满足。总之,随着基因组测序技术和高通量测序数据处理方法的发展,实用性的要求将越来越高,更便捷、快速和经济的测序平台势必会更加适应基层卫生疾病控制人员和科研人员的需求。
志谢 中国疾病预防控制中心传染病预防控制所张雯博士在实验过程中给予帮助,特此志谢利益冲突 无
| [1] |
Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection[J]. Annu Rev Pathol, 2019, 14: 319-338. DOI:10.1146/annurev-pathmechdis-012418-012751 |
| [2] |
Schlaberg R, Chiu CY, Miller S, et al. Validation of metagenomic next-generation sequencing tests for universal pathogen detection[J]. Arch Pathol Lab Med, 2017, 141(6): 776-786. DOI:10.5858/arpa.2016-0539-RA |
| [3] |
Palacios G, Druce J, Du L, et al. A new arenavirus in a cluster of fatal transplant-associated diseases[J]. N Engl J Med, 2008, 358(10): 991-998. DOI:10.1056/NEJMoa073785 |
| [4] |
Xu BL, Liu LC, Huang XY, et al. Metagenomic analysis of fever, thrombocytopenia and leukopenia syndrome (FTLS) in Henan province, China: discovery of a new bunyavirus[J]. PLoS Pathog, 2011, 7(11): e1002369. DOI:10.1371/journal.ppat.1002369 |
| [5] |
叶福强, 张锦海, 汪春晖. 纳米孔测序技术在病原体现场快速确认中的应用与挑战[J]. 中华卫生杀虫药械, 2019, 25(4): 374-378. Ye FQ, Zhang JH, Wang CH. Application and challenge of nanopore sequencing technology in rapid identification of pathogens in the field[J]. Chin J Hyg Insect Equip, 2019, 25(4): 374-378. DOI:10.19821/j.1671-2781.2019.04.021 |
| [6] |
Castro-Wallace SL, Chiu CY, John KK, et al. Nanopore DNA sequencing and genome assembly on the international space station[J]. Sci Rep, 2017, 7(1): 18022. DOI:10.1038/s41598-017-18364-0 |
| [7] |
Kilianski A, Haas JL, Corriveau EJ, et al. Bacterial and viral identification and differentiation by amplicon sequencing on the MinION nanopore sequencer[J]. GigaScience, 2015, 4(1): 12. DOI:10.1186/s13742-015-0051-z |
| [8] |
Hoenen T, Groseth A, Rosenke K, et al. Nanopore sequencing as a rapidly deployable Ebola outbreak tool[J]. Emerg Infect Dis, 2016, 22(2): 331-334. DOI:10.3201/eid2202.151796 |
| [9] |
Kafetzopoulou LE, Pullan ST, Lemey P, et al. Metagenomic sequencing at the epicenter of the Nigeria 2018 Lassa fever outbreak[J]. Science, 2019, 363(6422): 74-77. DOI:10.1126/science.aau9343 |
| [10] |
Quick J, Grubaugh ND, Pullan ST, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples[J]. Nat Protoc, 2017, 12(6): 1261-1276. DOI:10.1038/nprot.2017.066 |
| [11] |
Hansen S, Dill V, Shalaby MA, et al. Serotyping of foot-and-mouth disease virus using oxford nanopore sequencing[J]. J Virol Methods, 2019, 263: 50-53. DOI:10.1016/j.jviromet.2018.10.020 |
| [12] |
Moon J, Jang Y, Kim N, et al. Diagnosis of Haemophilus influenzae pneumonia by nanopore 16S amplicon sequencing of sputum[J]. Emerg Infect Dis, 2018, 24(10): 1944-1946. DOI:10.3201/eid2410.180234 |
| [13] |
Moon J, Kim N, Kim TJ, et al. Rapid diagnosis of bacterial meningitis by nanopore 16S amplicon sequencing: A pilot study[J]. Int J Med Microbiol, 2019, 309(6): 151338. DOI:10.1016/j.ijmm.2019.151338 |
| [14] |
Tessler M, Neumann JS, Afshinnekoo E, et al. Large-scale differences in microbial biodiversity discovery between 16S amplicon and shotgun sequencing[J]. Sci Rep, 2017, 7(1): 6589. DOI:10.1038/s41598-017-06665-3 |
| [15] |
Rao HX, Li SJ, Lu L, et al. Genetic diversity of Bartonella species in small mammals in the Qaidam Basin, western China[J]. Sci Rep, 2021, 11(1): 1735. DOI:10.1038/s41598-021-81508-w |
| [16] |
Weisburg WG, Barns SM, Pelletier DA, et al. 16S ribosomal DNA amplification for phylogenetic study[J]. J Bacteriol, 1991, 173(2): 697-703. DOI:10.1128/jb.173.2.697-703.1991 |
| [17] |
Zeng YH, Koblížek M, Li YX, et al. Long PCR-RFLP of 16S-ITS-23S rRNA genes: A high-resolution molecular tool for bacterial genotyping[J]. J Appl Microbiol, 2013, 114(2): 433-447. DOI:10.1111/jam.12057 |
| [18] |
Klindworth A, Pruesse E, Schweer T, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies[J]. Nucleic Acids Res, 2013, 41(1): e1. DOI:10.1093/nar/gks808 |
| [19] |
马洁琼, 栗冬梅, 陈忠科, 等. 鼠传巴尔通体流行概况[J]. 疾病监测, 2018, 33(1): 7-14. Ma JQ, Li DM, Chen ZK, et al. Epidemiological characteristics of rodent-borne Bartonella[J]. Dis Surveill, 2018, 33(1): 7-14. DOI:10.3784/j.issn.1003-9961.2018.01.004 |
| [20] |
Serratrice J, Rolain JM, Granel B, et al. Bilateral retinal artery branch occlusions revealing Bartonella grahamii infection[J]. Rev Med Interne, 2003, 24(9): 629-630. DOI:10.1016/s0248-8663(03)00224-8 |
| [21] |
Kerkhoff FT, Bergmans AMC, van Der Zee A, et al. Demonstration of Bartonella grahamii DNA in ocular fluids of a patient with neuroretinitis[J]. J Clin Microbiol, 1999, 37(12): 4034-4038. DOI:10.1128/JCM.37.12.4034-4038.1999 |
| [22] |
Mexas AM, Hancock SI, Breitschwerdt EB. Bartonella henselae and B. elizabethae as potential canine pathogens[J]. J Clin Microbiol, 2002, 40(12): 4670-4674. DOI:10.1128/jcm.40.12.4670-4674.2002 |
| [23] |
Julieta C, Amairani MR, Sonia TC, et al. First report of bacillary angiomatosis by Bartonella elizabethae in an HIV-positive patient[J]. Am J Dermatopathol, 2019, 41(10): 750-753. DOI:10.1097/DAD.0000000000001439 |
| [24] |
Cuscó A, Catozzi C, Vi es J, et al. Microbiota profiling with long amplicons using Nanopore sequencing: full-length 16S rRNA gene and whole rrn operon[J]. F1000Research, 2018, 7: 1755. DOI:10.12688/f1000research.16817.1 |
| [25] |
Meerburg BG, Singleton GR, Kijlstra A. Rodent-borne diseases and their risks for public health[J]. Crit Rev Microbiol, 2009, 35(3): 221-270. DOI:10.1080/10408410902989837 |
| [26] |
刘宁伟, 刘威, 黄留玉. 多重核酸检测技术研究进展[J]. 生物技术通讯, 2016, 27(4): 596-600. Liu NW, Liu W, Huang LY. Advances in research on nucleic acid-based multiplex detection technologies[J]. Lett Biotechnol, 2016, 27(4): 596-600. DOI:10.3969/j.issn.1009-0002.2016.04.031 |
| [27] |
李颖, 麻锦敏. 宏基因组学测序技术在中重症感染中的临床应用专家共识(第一版)[J]. 中华危重病急救医学, 2020, 32(5): 531-536. Li Y, Ma JM. Expert consensus for the application of metagenomic next generation sequencing in the pathogen diagnosis in clinical moderate and severe infections (first edition)[J]. Chin Crit Care Med, 2020, 32(5): 531-536. DOI:10.3760/cma.j.cn121430-20200228-00095 |
| [28] |
Quince C, Walker AW, Simpson JT, et al. Shotgun metagenomics, from sampling to analysis[J]. Nat Biotechnol, 2017, 35(9): 833-844. DOI:10.1038/nbt.3935 |
| [29] |
Tagini F, Greub G. Bacterial genome sequencing in clinical microbiology: A pathogen-oriented review[J]. Eur J Clin Microbiol Infect Dis, 2017, 36(11): 2007-2020. DOI:10.1007/s10096-017-3024-6 |
| [30] |
Mcnaughton AL, Roberts HE, Bonsall D, et al. Illumina and Nanopore methods for whole genome sequencing of hepatitis B virus (HBV)[J]. Sci Rep, 2019, 9(1): 7081. DOI:10.1038/s41598-019-43524-9 |
| [31] |
Koskela KA, Kalin-M ntt ri L, Hemmila H, et al. Metagenomic evaluation of bacteria from voles[J]. Vector Borne Zoonotic Dis, 2017, 17(2): 123-133. DOI:10.1089/vbz.2016.1969 |
| [32] |
Tiller RV, Gee JE, Frace MA, et al. Characterization of novel Brucella strains originating from wild native rodent species in North Queensland, Australia[J]. Appl Environ Microbiol, 2010, 76(17): 5837-5845. DOI:10.1128/AEM.00620-10 |
| [33] |
武少卿, 刘日宏, 杨哲宇. 啮齿动物布鲁杆菌病的自然疫源性研究进展[J]. 疾病监测与控制, 2015, 9(4): 246-247. Wu SQ, Liu RH, Yang ZY. The research progress of the natural focus Brucellosis of rodent[J]. J Dis Monitor Control, 2015, 9(4): 246-247. |
| [34] |
Greninger AL, Naccache SN, Federman S, et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis[J]. Genome Med, 2015, 7: 99. DOI:10.1186/s13073-015-0220-9 |
| [35] |
Tyler AD, Mataseje L, Urfano CJ, et al. Evaluation of Oxford nanopore's MinION sequencing device for microbial whole genome sequencing applications[J]. Sci Rep, 2018, 8(1): 10931. DOI:10.1038/s41598-018-29334-5 |