 2020, Vol. 31
2020, Vol. 31扩展功能
文章信息
- 张景山, 龙政, 王紫鉴, 肖文静, 王群, 赵宏群
- ZHANG Jing-shan, LONG Zheng, WANG Zi-jian, XIAO Wen-jing, WANG Qun, ZHAO Hong-qun
- 高通量半自动化细菌全基因组序列测定体系的构建
- Establishment of a high-throughput semi-automatic bacterial whole-genome sequencing system
- 中国媒介生物学及控制杂志, 2020, 31(6): 735-737
- Chin J Vector Biol & Control, 2020, 31(6): 735-737
- 10.11853/j.issn.1003.8280.2020.06.024
-
文章历史
- 收稿日期: 2020-06-11




2 河北北方学院, 河北 张家口 075000
2 Hebei North University
在生命科学领域,全基因组测序(whole genome sequencing,WGS)技术是20世纪80年代PCR诞生以来最重大的技术进步[1]。在过去的10余年中,WGS在病原体的遗传进化研究中发挥了重要作用[2-3]。近几年来,WGS的成本迅速下降,同时测序速度和测序数据量都大幅提高,WGS逐步地从科学研究转向实际应用[4]。在公共卫生领域,WGS在传染病的诊断和治疗中都发挥了作用。WGS包括诸多环节,大体可以分成数据的获取和数据分析2个部分。数据的获取涉及核酸制备、测序文库的构建以及序列测定,其中测序文库的构建是最为繁琐且对操作者要求最高的环节[1]。为了提高工作效率,自动化逐渐在许多实验室的日常工作中得到应用[5]。在公共卫生实验室中,由于工作量的加大和专业人员的相对稀缺,自动化的工作流程也逐渐开始得到应用[6]。
对于细菌性传染病,无论在监测还是该传染病暴发调查中,病原细菌的甄别都是公共卫生实验室工作的重点之一。基于单核苷酸多态性(single nucleotide polymorphism,SNP)的分型方法以及基于全基因组序列WGS的分型方法是当今及今后一段时间内的主流方法,而WGS是当前获取SNP信息最便捷的方式[7]。使用上述2种方法进行菌株甄别时,数据库的作用至关重要。用于比对的数据库不仅需要涵盖公共数据库,还需要包含所在国家、省历史分离菌株的基因组数据,这些历史菌株基因组数据的获得可以通过委托商业化测序公司获取,也可以在各自实验室自行测序获取。在应对细菌性疾病暴发时,由于生物安全以及实效的要求,测序通常由公共卫生实验室完成。手工文库构建操作繁琐且占用工作人员大量时间,因此,建立具有一定自动化程度高效的细菌基因组测序体系,对于常规监测和应对细菌性传染病暴发均具有重要意义。赵宏群等[6]的研究中建立了高通量、半自动化的细菌基因组核酸制备体系,能够在6 h内完成96个细菌样品的核酸提取与纯化。本研究在此基础上增加了自动测序文库构建系统,并与WGS测序仪组成了高通量、半自动化的细菌基因组新一代序列测定体系,该体系能够在55 h内完成96个样本的序列测定。
1 材料与方法 1.1 主要试剂及仪器96孔板型核酸提取与纯化试剂盒(69581,Qiagen,货号:145047553),蛋白酶K(Proteinase K,Amresco,货号:8019B073),RNA酶A(RNase A,Promega,货号:0000118628),MGIEasy酶切DNA文库制备试剂套装(MGI,货号:1000017572),Qubit dsDNA HS Assay Kit (Invitrogen,批号:Q32854),安捷伦高灵敏度DNA分析试剂盒(Agilent,货号:5067-4626),安捷伦DNA分析试剂盒(Agilent,货号:5067-1504),96孔PCR板(Bio-Rad,LE2001);丢弃型机械手枪头(RoboRack MDT 235,PerkinElmer,6001295及RoboRack MDT 25,PerkinElmer,6000677),自动化液体处理器(Janus,PerkinElmer,8通道及96通道),微量分光光度计(NanoDrop 1000,NanoDrop),温控孵育模块(Inheco multi TEC control,8900030),Qubit4.0荧光定量仪(ThermoFisher,货号:Q33216),Agilent 2100 Bioanalyzer (Agilent Technologies,货号:G2939AA),MGISEQ-2000(PE150读长)。
1.2 核酸制备核酸制备按照赵宏群等[6]的研究方法进行。
1.3 自动化文库构建将放置样品的96孔板、250 μl带滤芯的丢弃型机械手枪头、96孔PCR板、MGIEasy酶切DNA文库制备试剂套装中的Frag Buffer、Frag Enzyme、ERAT Buffer、ERAT Enzyme Mix、Ligation Buffer、DNA Ligase、PCR Enzyme Mix、PCR Primer Mix、adaptor以及磁珠、80%乙醇溶液和TE分别置于自动化液体处理器的不同板位,经过DNA片段化、磁珠片段筛选、末端修复并添加dA尾、接头连接、连接产物纯化、PCR扩增和PCR产物纯化等步骤得到纯化后的PCR产物。纯化后的产物经过变性、酶切消化、产物纯化后,进行质检。合格的文库即可用于序列测定。
1.4 基因组序列测定使用华大基因制造的MGISEQ-2000测序仪,按照PE150方式进行基因组序列测定。
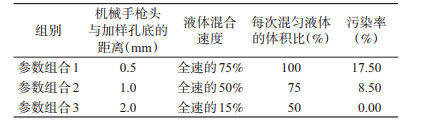
2 结果 2.1 文库构建中不同参数组合与样品交叉污染率比较我们测定了机械手枪头与加样孔底的距离、液体混合速度和每次混匀液体的体积比(每次吸入液体/总液体)这3个因素对交叉污染的影响。分别固定2个参数,只改变第3个参数,比较这3个参数对不同样品孔之间污染率的影响,得到3个交叉污染率最小的参数组合(表 1),其中参数组合3的污染率为0。
|
为了降低自动化液体处理器的使用成本,对文库构建过程中使用可丢弃型机械手枪头的步骤进行研究发现,在所有涉及可丢弃型机械手枪头的步骤中,DNA片段化、磁珠片段筛选、末端修复并添加dA尾、接头连接和连接产物纯化这些步骤的DNA浓度较低(20 ng/μl),因此,在这些步骤中未更换机械手枪头,而是通过使用次氯酸溶液和超纯水交替洗涤3次的方法解决DNA交叉污染的问题。在每次进行涉及DNA的操作之后,使用200 μl的浓度为200 mg/L的次氯酸冲洗枪头1次,然后用200 μl的超纯水冲洗1次,此过程重复3次。之后,外加超纯水重新冲洗3次的方法洗涤机械手枪头。在奇数列的奇数行所有孔中均加入纯化后的大肠埃希菌基因组核酸,在剩余的所有孔中只加入纯水,使用上述冲洗方案洗涤机械手枪头构建文库,对构建的文库进行测序分析,结果在只加入纯水的样品孔中未检测到大肠埃希菌基因组特异的核酸序列。因此,通过本方案的实施,减少了50%可丢弃型机械手枪头的使用量。由于增加了洗涤可丢弃型机械手枪头的步骤,整个操作过程约增加0.5 h。
3 讨论无论在临床还是公共卫生实验室中,新一代基因组测序(NGS)均得到越来越广泛地应用。我国的国家致病菌识别网(pathogen identification net,PIN)及美国疾病预防与控制中心的高级分子检测项目均已经开始将基因组数据应用到监测和/或检测中[1]。从获取样本到得到NGS分析结果这个流程中,目前至少存在2个瓶颈,分别是测序文库构建和数据分析。在大多数公共卫生实验室中,目前的测序文库构建主要依赖人工完成,存在操作繁琐、时间长等缺点。因此,本研究在前期工作的基础上[6],尝试使用自动化液体工作站着重解决公共卫生实验室中测序文库构建的问题。
文库构建中涉及大量的液体混匀操作,容易产生气溶胶,气溶胶是不同样品文库之间污染的最主要来源。因此,在构建测序文库体系时,我们比较了可丢弃型机械手枪头与加样孔底的距离、液体混合速度和每次混匀液体的体积比对样品的交叉污染率的影响。结果显示,可丢弃型机械手枪头与加样孔底的距离越小,液体混合速度越快; 每次混匀液体的体积比例越大,样品之间的交叉污染率越高; 反之,交叉污染率则降低。这是因为,液体混合速度越快,形成的冲击力就越大,相应地产生气溶胶的可能性就越大; 每次混匀液体的体积比例越高,则形成的冲击液体体积就越大,产生气溶胶的可能性也就相应地增大; 而可丢弃型机械手枪头与加样孔底之间的距离越小,则容易形成气溶胶,形成的气溶胶会从样品孔中扩散到相邻的样品孔中,从而导致样品之间以及文库之间的交叉污染。
在保证样品之间不发生交叉污染的情况下最大限度地减少可丢弃型机械手枪头的使用,我们还尝试解决半自动化过程中可丢弃型机械手枪头过多、导致自动化液体工作站运行成本过高的问题。研究结果提示,在DNA片段化、磁珠片段筛选、末端修复并添加dA尾、接头连接和连接产物纯化这些步骤中,使用次氯酸溶液和超纯水洗涤3次、外加超纯水冲洗3次机械手枪头的方法能够避免样品之间的交叉污染。该洗涤步骤虽然延长了工作时间(约0.5 h),但是却减少了25%可丢弃型机械手枪头的使用量,从而降低了整个体系的运行成本,有利于该体系的应用。
综上所述,本研究建立的高通量半自动化细菌全基因组序列测定体系,能够在55 h内完成从核酸制备到数据产出的过程,使用优化后的洗涤可丢弃型机械手枪头,能够在避免样品之间交叉污染的情况下节省25%的可丢弃型机械手枪头的使用量; 与手工方法相比,该体系能够提高工作效率4倍。随着测序文库新方案的出现,文库构建的时间会进一步缩短,相应地使用成本也会随之下降,从而有利于本方案的推广使用。
| [1] |
Gargis AS, Kalman L, Lubin IM. Assuring the quality of next-generation sequencing in clinical microbiology and public health laboratories[J]. J Clin Microbiol, 2016, 54(12): 2857-2865. DOI:10.1128/JCM.00949-16 |
| [2] |
Morelli G, Song YJ, Mazzoni CJ, et al. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity[J]. Nat Genet, 2010, 42(12): 1140-1143. DOI:10.1038/ng.705 |
| [3] |
Didelot X, Pang B, Zhou ZM, et al. The role of china in the global spread of the current cholera pandemic[J]. PLoS Genet, 2015, 11(3): e1005072. DOI:10.1371/journal.pgen.1005072 |
| [4] |
Pang B, Du PC, Zhou ZM, et al. The transmission and antibiotic resistance variation in a multiple drug resistance clade of vibrio cholerae circulating in multiple countries in Asia[J]. PLoS One, 2016, 11(3): e0149742. DOI:10.1371/journal.pone.0149742 |
| [5] |
Blow N. Lab automation:tales along the road to automation[J]. Nat Methods, 2008, 5(1): 109-112. DOI:10.1038/nmeth0108-109 |
| [6] |
赵宏群, 卢昕, 逄波. 高通量半自动细菌核酸提取与纯化体系的构建[J]. 疾病监测, 2016, 31(3): 256-259. Zhao HQ, Lu X, Pang B. Establishment of high-throughput half-automated nucleotide extraction and purification system[J]. Dis Surveill, 2016, 31(3): 256-259. DOI:10.3784/j.issn.1003-9961.2016.03.017 |
| [7] |
Kan B, Zhou HJ, Du PC, et al. Transforming bacterial disease surveillance and investigation using whole-genome sequence to probe the trace[J]. Front Med, 2018, 12(1): 23-33. DOI:10.1007/s11684-017-0607-7 |