 2020, Vol. 31
2020, Vol. 31扩展功能
文章信息
- 刘芷涵, 孙小红, 邢一帆, 周丹, 孙艳, 马磊, 沈波
- LIU Zhi-han, SUN Xiao-hong, XING Yi-fan, ZHOU Dan, SUN Yan, MA Lei, SHEN Bo
- 淡色库蚊溴氰菊酯敏感品系与抗性种群的肠道菌群多样性分析
- An analysis of gut microbial diversity of deltamethrin-susceptible strains and deltamethrin-resistant populations of Culex pipiens pallens
- 中国媒介生物学及控制杂志, 2020, 31(5): 545-551
- Chin J Vector Biol & Control, 2020, 31(5): 545-551
- 10.11853/j.issn.1003.8280.2020.05.009
-
文章历史
- 收稿日期: 2020-04-27




2 江苏省现代病原学重点实验室, 江苏 南京 211166;
3 南京医科大学第一附属医院, 江苏 南京 211166
2 Key Laboratory of Pathogen Biology(NJMU);
3 First Affiliated Hospital of Nanjing Medical University
蚊虫属双翅目(Diptera)蚊科(Culicidae),是一类重要的医学昆虫,能通过吸血传播疟疾、登革热和黄热病等严重疾病,全球每年约有7亿人感染蚊媒病,死亡人数高达100万例,由于缺乏有效的治疗药物和疫苗,蚊媒控制是目前蚊媒病防治的主要策略。通过杀虫剂灭蚊能有效控制蚊媒数量,减少蚊媒病的传播,但杀虫剂广泛大量的使用促进了抗药性的发生发展,严重阻碍了蚊媒病的防治进程,因此,亟需对蚊虫抗药性机制进行深入研究。以往的抗药性机制研究主要围绕靶标位点突变[1]、解毒酶代谢水平上升[2-3]和表皮增厚减少渗透3个方面[4-5],而最近有学者发现昆虫肠道共生菌能在杀虫剂抗性中发挥重要作用[6-7]。
已知肠道菌群与昆虫宿主间的协同进化由来已久,昆虫的多样化和进化成功部分取决于它们与肠道菌群的多种联系。随着现代测序技术和分子生物学技术的进步,扩展了我们对肠道环境的生态以及菌群功能学的认知,逐渐挖掘出肠道菌群在昆虫宿主的生长发育[8]、昆虫肠道共生菌能在杀虫剂抗性中发挥重要作用交配偏好[9]、寿命以及对病原体的免疫防御等多方面具有重要作用,并且通过对昆虫肠道共生菌的操纵来进行害虫防治的相关研究部分已成功应用于实际。例如,通过利用苏云金杆菌产生的δ-内毒素蛋白晶体杀灭昆虫幼虫,以及基于生殖细胞胞质不相容技术(cytoplasmic incompatibility,CI)使雄性昆虫感染沃尔巴克氏体,导致交配后的雌性昆虫繁殖力大大降低等[10-11]。因此,只有通过了解昆虫的肠道菌群多样性和结构组成,才有利于操纵肠道共生菌从而控制害虫。然而迄今为止,鲜有研究对成蚊整体肠道微生物进行报道,鉴于昆虫肠道菌群的重要功能,有必要对蚊虫的肠道微生物进行研究,了解肠道微生物与蚊虫之间的相互作用和作用范围。本研究通过对16SrDNA基因V3+V4区的高通量测序,比较敏感品系与抗性种群肠道菌群多样性和组成结构差异,从而获得具有特殊功能的菌群资源,为进一步理解蚊虫与肠道细菌之间的协同进化和互作关系提供依据。
1 材料与方法 1.1 供试蚊虫淡色库蚊(Culex pipiens pallens)溴氰菊酯敏感品系于2012年8月采自江苏省南京市郊区水塘的淡色库蚊自然种群,未接触溴氰菊酯等杀虫剂进行传代饲养。淡色库蚊溴氰菊酯抗性种群为以敏感品系为起始种群,按世界卫生组织(WHO)幼虫浸渍法,用半数致死浓度(LC50)的溴氰菊酯对敏感品系逐代筛选获得。敏感品系与抗性种群的溴氰菊酯LC50分别为0.13和8.71 mg/L,抗性倍数为66.92倍。饲养条件为温度28 ℃,湿度70%~80%,光照时间为14 h/d。
1.2 淡色库蚊肠道的分离选择羽化3 d的敏感品系与抗性种群雌蚊共300只,饥饿处理6 h后,在75 %无菌乙醇溶液中浸泡3 min,以体表消毒,再用无菌磷酸盐缓冲液(PBS)清洗3次,无菌水清洗3次。在显微镜下分离解剖,然后将肠道转移到无菌1.5 ml离心管中,每管50个肠道,设置3个生物学重复。
1.3 淡色库蚊肠道总DNA提取和16S rDNA V3+V4区的PCR扩增使用DNA微量提取试剂盒〔天根生化科技(北京)有限公司〕提取上述1.2中收集的肠道样本的总细菌基因组DNA,并在1.0%的琼脂糖凝胶上电泳检测基因组DNA抽提质量。为了扩增用于高通量测序的16S rDNA,以通用引物338F/806R(5′-ACTCCTACGGAGGCAGCA-3′;5′-GGACTAC HVGGGTWTCTAAT-3′)对V3+V4区进行扩增。PCR的总体积为50 μl:ddH2O 19 μl,2×Vazyme Lamp Master Mix 25 μl,引物338F/806R各2 μl,模板DNA 2 μl。PCR的扩增条件:94 ℃ 5 min;94 ℃ 30 s,55 ℃ 30 s,72 ℃ 30 s,35个循环;72 ℃ 7 min。扩增产物在1.0%琼脂糖凝胶上电泳并纯化,然后将产物发送到上海美吉生物医药科技有限公司,以构建V3+V4文库进行测序。
1.4 16S rDNA基因V3+V4区深度测序和数据分析将测序获得的原始数据根据overlap关系进行拼接,并对序列质量进行质控过滤,去除低质量的序列以获得优化序列数据。根据Uparse软件对优化序列按照97%的序列相似性进行聚类,以获得目标操作分类单元(OTUs)。使用Silva参考数据库进行OTUs注释。通过QIIME 2.0软件生成物种丰度表,并使用R软件绘制每个分类阶元类别的群落结构。使用Mothur v1.30.2软件评估反映样本物种丰富度的Chao1指数和反映样本物种多样性的香农指数及辛普森指数的Alpha多样性指标。
1.5 测序结果实时荧光定量PCR(RT-qPCR)的验证随机挑选注释到的敏感品系与抗性种群间丰度差异较大的肠道菌进行验证,表 1中描述了用于RT-qPCR的引物序列。RT-qPCR总体系为20 μl:2×Eva Green qPCR Master Mix-No Dye 10 μl,正、反向引物(10 mmol/L)各1 μl,模板DNA 8 μl。RT-qPCR的扩增条件:95 ℃ 600 s,95 ℃ 15 s,60 ℃ 60 s,共40个循环。在RT-qPCR结束后进行溶解曲线分析,评估扩增引物的特异性。

采用GraphPad Prism 8软件进行数据处理和分析,所有数据均采用平均数±标准差(x±s)表示,采用双尾t检验比较两组之间的差异。P<0.05为差异有统计学意义。
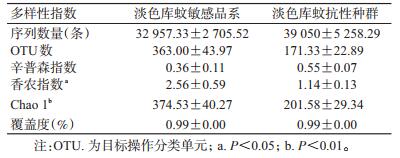
2 结果 2.1 测序质量统计与OTUs分类通过Illumina MiSeq高通量测序对敏感品系与抗性种群肠道细菌16S rDNA基因V3+V4可变区进行测序分析,共产生435 843条原始序列,经过两端序列拼接优化后得到216 022条有效序列。样本中99.99%的序列在400~600 bp之间,平均长度为429 bp。按照Classifier贝叶斯算法将它们以97%的序列相似性进行聚类分析,获取所有样本的OTUs数量概况,共获得766个OTUs,其中258个为敏感品系与抗性种群所共有,分别有472和36个为敏感品系及抗性种群独有(图 1)。

|
| 图 1 淡色库蚊敏感品系与抗性种群肠道的OTUs分布维恩图 Figure 1 V enn diagram of operational taxonomic unit distribution in the gut of deltamethrin-susceptible strains and deltamethrin-resistant populations of Culex pipiens pallens |
| |
为了衡量敏感品系与抗性种群肠道菌群的多样性和群落的物种丰富度,对Alpha多样性指数(辛普森指数、香农指数、Chao1指数及覆盖度)数据进行统计分析(表 2),OTUs数、Chao1指数越高,说明群落总数及丰富度越高;香农指数越高,辛普森指数越低,说明物种多样性越高。根据覆盖度显示,敏感品系与抗性种群肠道菌群的平均采样覆盖率接近100%,说明大多细菌都已检出,较少细菌未检测到。而敏感品系的香农指数和Chao1指数均显著高于抗性种群,差异有统计学意义(t=4.072,df=4,P=0.015;t=6.012,df=4,P=0.003)。
通过随机抽取测序序列与其所代表的OTU数目绘制稀释曲线来分析测序数据的饱和程度(图 2),结果显示,随着测序深度加深,敏感品系肠道测序的稀释曲线趋于平缓并接近饱和,表明获得的测序量能够客观反映敏感品系肠道的所有细菌类群;抗性种群的稀释曲线在97%相似水平下曲线斜率逐渐降低,但尚未完全进入平台期,说明增加测序量会发现少量新的OTUs。通过Rank-Abundance曲线来反映敏感品系与抗性种群肠道菌群的物种丰富度和均匀度。物种丰富度由曲线在横轴上的长度来反映,曲线越宽则物种丰富度越高;物种均匀度由曲线的形状来反映,曲线越平坦则物种组成的均匀度越高。如图 2所示,敏感品系与抗性种群在垂直方向的平滑度较低,水平方向上跨度很大,说明敏感品系与抗性种群肠道菌群的组成较丰富,但抗性种群的物种分布均匀度不高,说明样本中存在占比较高的优势菌群。通过PAST 1.0软件分析敏感品系与抗性种群肠道菌群的Beta多样性,如非度量多维标度图(NMDS)所示(图 3),横轴和纵轴仅其图示作用,抗性种群分布在图左区域,敏感品系分布在图右区域,不同细菌群落NMDS分析表明敏感品系与抗性种群肠道细菌群落构成差异有统计学意义(stress=0)。

|
| 注:OTU.表示目标操作分类单元。 图 2 稀释曲线和Rank-abundance曲线 Figure 2 Rarefaction curves and Rank-abundance curves |
| |

|
| 注:DS.为敏感品系; DR.为抗性种群。 图 3 非度量多维标度(NMDS)图 Figure 3 Non-metric multidimensional scaling (NMDS) plot |
| |
根据OTUs的注释结果显示,敏感品系与抗性种群的肠道菌群共获得31个门,62个纲,132个目,215个科和339个属。在门分类阶元水平上(图 4A),主要注释了变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)、放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)及拟杆菌门(Bacteroidetes)等31个门。变形菌门在抗性种群中占绝对优势,相对丰度为(96.52±1.74)%,敏感品系的优势菌门与之相同,但其相对丰度为(79.30±5.13)%,显著低于抗性种群,差异有统计学意义(t=5.501,df=4,P=0.005)。次优势菌门均为厚壁菌门,其在敏感品系与抗性种群中相对丰度分别为(3.71±0.28)%和(1.33±1.61)%,差异无统计学意义(t=2.520,df=4,P=0.065)。上述2个门在敏感品系与抗性种群的累积丰度分别为82.98%和97.83%,剩余29个门在敏感品系与抗性种群的累积丰度仅为17.02%和2.17%。

|
| 图 4 敏感品系与抗性种群肠道菌群相对丰度 Figure 4 Relative abundance of gut microbiota of deltamethrin- suscept tible strains and deltamethrin-resistant populations |
| |
在目分类阶元水平上(图 4B),敏感品系中肠道菌群相对丰度最高的前3位依次为红螺菌目(Rhodospirillales)、肠杆菌目(Enterobacteriles)及伯克霍尔德氏菌目(Burkholderiales),其相对丰度分别为(65.92±8.94)%、(3.20±1.17)%和(2.68±0.47)%;而抗性种群中相对丰度最高的前3位为红螺菌目、气单胞菌目(Aeromonadales)及乳酸杆菌目(Lactobacillales),相对丰度分别为(87.42±5.89)%、(7.07±6.70)%和(1.05±1.51)%,其中敏感品系的肠杆菌目(t=3371,df=4,P=0.028)、伯克霍尔德氏菌目(t=7.975,df=4,P=0.001)、梭菌目(Clostridiales)(t=3.471,df=4,P=0.025)和根瘤菌目(Rhizobiales)(t=3.547,df=4,P=0.023)的相对丰度显著高于抗性种群,差异有统计学意义,而抗性种群的红螺菌目相对丰度显著高于敏感品系,差异有统计学意义(t=3.479,df=4,P=0.025)。
在属分类阶元水平上(图 4C),敏感品系中肠道菌群相对丰度>1%的菌属主要为未鉴定属的醋酸杆菌科(Acetobacteraceae)、朝井杆菌属(Asaia)、红球菌属(Rhnodococcus)、罗尔斯通氏菌属(Ralstonia)、大肠志贺杆菌属(Escherichia-Shigella)、梭菌属(Clostrudium)、未鉴定属的酸杆菌门(Acidobacteria)和克雷伯氏菌属(Klebsiella)。其中未鉴定属的醋酸杆菌科(Acetobacteraceae)作为敏感品系的优势菌群占比为(60.05±8.83)%,其相对丰度显著高于抗性种群,差异有统计学意义(t=9.687,df=4,P=0.001)。敏感品系的次优势菌群分别为朝井杆菌属和红球菌属(Rhnodococcus),占比分别为(5.37±1.77)%和(2.13±1.23)%,其中朝井杆菌属的相对丰度远低于抗性种群,差异有统计学意义(t=21.82,df=4,P=0.001)。相比于敏感品系,朝井杆菌属(76.94±5.40)%、未鉴定属的醋酸杆菌科(10.47±0.76)%、气单胞属(Aeromonas)(7.07±6.70)%和肠球菌属(Enterococcus)(1.07±0.15)%以95.40%的累计丰度占据抗性种群肠道,剩余菌属丰度仅为4.60%。
2.4 高通量测序结果的RT-qPCR验证随机选取敏感品系与抗性种群肠道中相对丰度具有较大差异的细菌朝井杆菌属,并基于测序信息进行特异性引物设计,通过RT-qPCR技术来验证高通量测序数据的可靠性(图 5)。结果显示,溶解曲线为单一峰,证实所设计引物特异性较好,抗性种群肠道内朝井杆菌属的相对丰度显著高于敏感品系,且差异有统计学意义(t=3.043,df=4,P=0.038),与测序结果一致。
|
|
| 注:图中数据为3次重复的平均值+标准差; a. P=0.038。 图 5 高通量测序结果的实时荧光定量PCR验证结果 Figure 5 Verification results of the high-throughput sequencing by real-time fluorescent quantitative PCR |
| |
本研究通过Illumina MiSeq高通量测序技术对淡色库蚊敏感品系与抗性种群的肠道细菌群落进行分析,共注释到了31个门,62个纲,132个目,215个科和339个属。变形菌门和厚壁菌门是淡色库蚊敏感品系与抗性种群肠道中的主要构成菌门,该结果与多数昆虫肠道的优势菌相似,如膜翅目的中华蜜蜂(Apis cerana)[12]、切叶蚁(Acromyrmex)[13]、半翅目的褐飞虱(Nilaparvata lugens)[14-15]、温带臭虫(Cimex lectularius)[16]以及双翅目的果蝇(Drosophila)[17]等,然而其相对丰度在不同昆虫中存在一定的差异。昆虫肠道中普遍检测到变形菌门细菌的高丰度分布,提示昆虫肠道环境对变形菌门细菌的选择性以及适应进化,同时变形菌门细菌也为昆虫的生理活动发挥作用。
昆虫在进化过程中,肠道微生物通过与宿主的互惠共生,协同进化,促进和提高了昆虫的生命活动。在淡色库蚊敏感品系与抗性种群肠道中鉴定的132个目和339个属中,其中许多细菌可能参与了淡色库蚊的生命活动。肠杆菌目、不动杆菌目及根瘤菌目等可能参与淡色库蚊肠道营养物质代谢。肠杆菌目能够产生溶血酶,促进吸血后食物的消化;不动杆菌目能够促进碳氮的代谢,具有氮素转化的功能;根瘤菌目能产生具有纤维素和果胶水解活性的酶,帮助宿主合成含氮物质。
除了参与宿主的营养与代谢,在敏感品系与抗性种群肠道中发现了许多可能与有毒物质降解有关的菌属,如红球菌属、肠球菌属、克雷伯氏菌属等。红球菌属的细菌能分泌对昆虫有害的单萜烯类物质的酶[18];肠球菌属能产生乙酸盐,降低肠道的pH值,保护昆虫宿主免受毒素的伤害;克雷伯氏菌属能将有机氯杀虫剂硫丹降解为硫丹内酯[19]。此外,有些肠道细菌还能够参与宿主的免疫与防御,敏感品系与抗性种群肠道中的大肠志贺杆菌属、假单胞杆菌属、朝井杆菌属可能参与蚊的免疫反应。有报道指出,当家蚕口饲大肠志贺杆菌属时,葛佬素和溶菌酶基因表达上升,而口饲假单胞杆菌属时,葛佬素2和葛佬素3以及免疫相关通路-9的表达上升[20],说明这2种肠道菌与家蚕的免疫信号通路有着密切的关系。而朝井杆菌属能够激活抗菌肽的表达促进蚊的免疫防御,减少蚊虫对疟疾的感染。肠道菌群在影响宿主生理活动的同时,昆虫肠道菌群结构也受到了环境、饮食等因素的制约。敏感品系与抗性种群肠道中都含有较高丰度的未鉴定属醋酸杆菌科,这可能与它们高糖饮食有关。因淡色库蚊成蚊主要以花蜜为食,而醋酸杆菌科细菌广泛分布于植物花朵、根茎以及软饮料、食醋等发酵食品中,在以花蜜为基础饮食的双翅目[21]、膜翅目[22]、鳞翅目[23]及半翅目[24]昆虫肠道中均有较高丰度。
昆虫的肠道菌群组成结构还会受到理化条件的影响。在本研究中,基于对淡色库蚊敏感品系与抗性种群肠道菌群的比较分析,结果发现经杀虫剂逐代筛选的有共同起源的淡色库蚊敏感品系和抗性种群肠道细菌种类丰富,但敏感品系肠道细菌的物种总数和种类多样性高于抗性种群肠道细菌,两者的肠道菌群包含258个共有的OTU,又分别具有472和36个独特的OTU。与敏感品系比较,抗性种群肠道内具有高丰度的朝井杆菌属和气单胞菌属,累计丰度高达83.84%,而相应的红球菌属、罗尔斯通氏菌属、芽单胞菌属及酸微菌属的丰度较低。肠道菌群对杀虫剂的响应提示杀虫剂的接触会对淡色库蚊的肠道菌群具有选择作用,在杀虫剂世代暴露的选择压力时肠道能富集具有竞争优势的菌群,而这些独特的OTU可能在蚊的杀虫剂抗性中产生作用。这类因杀虫剂的暴露导致昆虫肠道菌群组成结构的改变先前已有研究报道,如经2种不同有机磷杀虫剂世代接触筛选后的小菜蛾肠道内乳杆菌的丰度显著增加[18];将对硫磷喷洒在土壤中,会导致土壤内具有对硫磷降解能力的伯克氏菌属大量富集[25];以及在用螺旋藻处理后的蚜虫,其肠道菌群也发生了类似变化[26]。2015年有研究报道,具有杀虫剂降解能力的细菌,如棒状杆菌属(降解有机磷)、红球菌属(降解氯氰菊酯)和芽孢杆菌属(降解茚虫威)等,在本研究的敏感品系与抗性种群中也有发现,但所占丰度较小,这些属具备杀虫剂降解能力是否参与了淡色库蚊的杀虫剂抗性还有待进一步研究验证,但昆虫肠道菌群可能参与杀虫剂的降解代谢。这些结果提示,昆虫肠道菌群结构的差异对于昆虫宿主具有重要作用,淡色库蚊敏感品系与抗性种群肠道细菌的差异是否与杀虫剂抗性有关,值得进一步实验探索。
本研究基于Illumina MiSeq测序揭示了淡色库蚊敏感品系与抗性种群肠道菌群的多样性和组成结构,分析了淡色库蚊肠道菌群参与营养代谢、有毒物质解毒和宿主的免疫防御等功能。淡色库蚊敏感品系与抗性种群肠道菌群结构的差异,提示肠道菌群的变化可能在杀虫剂抗性中发挥重要作用。通过对淡色库蚊肠道菌群的研究,有助于深入理解蚊虫及肠道菌群间的协同进化和互作关系,其与杀虫剂抗性的关联为蚊媒的综合防治提供新的视角和线索。
| [1] |
Kawada H, Higa Y, Futami K, et al. Discovery of point mutations in the voltage-gated sodium channel from African Aedes aegypti populations:potential phylogenetic reasons for gene introgression[J]. PLoS Negl Trop Dis, 2016, 10(6): e0004780. DOI:10.1371/journal.pntd.0004780 |
| [2] |
Grigoraki L, Lagnel J, Kioulos I, et al. Transcriptome profiling and genetic study reveal amplified carboxylesterase genes implicated in temephos resistance, in the Asian tiger mosquito Aedes albopictus[J]. PLoS Negl Trop Dis, 2015, 9(5): e0003771. DOI:10.1371/journal.pntd.0003771 |
| [3] |
Leong CS, Vythilingam I, Liew JWK, et al. Enzymatic and molecular characterization of insecticide resistance mechanisms in field populations of Aedes aegypti from Selangor, Malaysia[J]. Parasit Vectors, 2019, 12(1): 236. DOI:10.1186/s13071-019-3472-1 |
| [4] |
Hemingway J, Hawkes NJ, McCarroll L, et al. The molecular basis of insecticide resistance in mosquitoes[J]. Insect Biochem Mol Biol, 2004, 34(7): 653-665. DOI:10.1016/j.ibmb.2004.03.018 |
| [5] |
Tabbabi A, Daaboub J, Cheikh RB, et al. Resistance status to deltamethrin pyrethroid of Culex pipiens pipiens (Diptera:Culicidae) collected from three districts of Tunisia[J]. Afr Health Sci, 2018, 18(4): 1182-1188. DOI:10.4314/ahs.v18i4.39 |
| [6] |
Pietri JE, Tiffany C, Liang DS. Disruption of the microbiota affects physiological and evolutionary aspects of insecticide resistance in the German cockroach, an important urban pest[J]. PLoS One, 2018, 13(12): e0207985. DOI:10.1371/journal.pone.0207985 |
| [7] |
Cheng DF, Guo ZJ, Riegler M, et al. Gut symbiont enhances insecticide resistance in a significant pest, the oriental fruit fly Bactrocera dorsalis (Hendel)[J]. Microbiome, 2017, 5(1): 13. DOI:10.1186/s40168-017-0236-z |
| [8] |
Ben-Yosef M, Pasternak Z, Jurkevitch E, et al. Symbiotic bacteria enable olive flies (Bactrocera oleae) to exploit intractable sources of nitrogen[J]. J Evol Biol, 2014, 27(12): 2695-2705. DOI:10.1111/jeb.12527 |
| [9] |
Dan H, Ikeda N, Fujikami M, et al. Behavior of bacteriome symbionts during transovarial transmission and development of the Asian citrus psyllid[J]. PLoS One, 2017, 12(12): e0189779. DOI:10.1371/journal.pone.0189779 |
| [10] |
Beckmann JF, Ronau JA, Hochstrasser M. A Wolbachia deubiquitylating enzyme induces cytoplasmic incompatibility[J]. Nat Microbiol, 2017, 2(5): 17007. DOI:10.1038/nmicrobiol.2017.7 |
| [11] |
Landmann F. The Wolbachia endosymbionts[J]. Microbiol Spectr, 2019, 7(2). DOI:10.1128/microbiolspec.BAI-0018-2019 |
| [12] |
Ahn JH, Hong IP, Bok JI, et al. Pyrosequencing analysis of the bacterial communities in the guts of honey bees Apis cerana and Apis mellifera in Korea[J]. J Microbiol, 2012, 50(5): 735-745. DOI:10.1007/s12275-012-2188-0 |
| [13] |
Zhukova M, Sapountzis P, Schiott M, et al. Diversity and transmission of gut bacteria in Atta and Acromyrmex leaf-cutting ants during development[J]. Front Microbiol, 2017, 8: 1942. DOI:10.3389/fmicb.2017.01942 |
| [14] |
Douglas AE. Symbiotic microorganisms:untapped resources for insect pest control[J]. Trends Biotechnol, 2007, 25(8): 338-342. DOI:10.1016/j.tibtech.2007.06.003 |
| [15] |
Narasimhan S, Fikrig E. Tick microbiome:the force within[J]. Trends Parasitol, 2015, 31(7): 315-323. DOI:10.1016/j.pt.2015.03.010 |
| [16] |
Meriweather M, Matthews S, Rio R, et al. A 454 survey reveals the community composition and core microbiome of the common bed bug (Cimex lectularius) across an urban landscape[J]. PLoS One, 2013, 8(4): e61465. DOI:10.1371/journal.pone.0061465 |
| [17] |
Broderick NA, Lemaitre B. Gut-associated microbes of Drosophila melanogaster[J]. Gut Microbes, 2012, 3(4): 307-321. DOI:10.4161/gmic.19896 |
| [18] |
Xia XF, Zheng DD, Zhong HZ, et al. DNA sequencing reveals the midgut microbiota of diamondback moth, Plutella xylostella (L.) and a possible relationship with insecticide resistance[J]. PLoS One, 2013, 8(7): e68852. DOI:10.1371/journal.pone.0068852 |
| [19] |
Kwon GS, Sohn HY, Shin KS, et al. Biodegradation of the organochlorine insecticide, endosulfan, and the toxic metabolite, endosulfan sulfate, by Klebsiella oxytoca KE-8[J]. Appl Microbiol Biotechnol, 2005, 67(6): 845-850. DOI:10.1007/s00253-004-1879-9 |
| [20] |
Douglas AE. The B vitamin nutrition of insects:the contributions of diet, microbiome and horizontally acquired genes[J]. Curr Opin Insect Sci, 2017, 23: 65-69. DOI:10.1016/j.cois.2017.07.012 |
| [21] |
Crotti E, Damiani C, Pajoro M, et al. Asaia, a versatile acetic acid bacterial symbiont, capable of cross-colonizing insects of phylogenetically distant genera and orders[J]. Environ Microbiol, 2009, 11(12): 3252-3264. DOI:10.1111/j.1462-2920.2009.02048.x |
| [22] |
Good AP, Gauthier MPL, Vannette RL, et al. Honey bees avoid nectar colonized by three bacterial species, but not by a yeast species, isolated from the bee gut[J]. PLoS One, 2014, 9(1): e86494. DOI:10.1371/journal.pone.0086494 |
| [23] |
Robinson CJ, Schloss P, Ramos Y, et al. Robustness of the bacterial community in the cabbage white butterfly larval midgut[J]. Microb Ecol, 2010, 59(2): 199-211. DOI:10.1007/s00248-009-9595-8 |
| [24] |
Gonella E, Crotti E, Rizzi A, et al. Horizontal transmission of the symbiotic bacterium Asaia sp. in the leafhopper Scaphoideus titanus ball (Hemiptera:Cicadellidae)[J]. BMC Microbiol, 2012, 12 Suppl 1: S4. DOI:10.1186/1471-2180-12-s1-s4 |
| [25] |
Kikuchi Y, Hayatsu M, Hosokawa T, et al. Symbiont-mediated insecticide resistance[J]. Proc Natl Acad Sci USA, 2012, 109(22): 8618-8622. DOI:10.1073/pnas.1200231109 |
| [26] |
Zhang J, Pan YO, Zheng C, et al. Rapid evolution of symbiotic bacteria populations in spirotetramat-resistant Aphis gossypii glover revealed by pyrosequencing[J]. Comp Biochem Physiol Part D Genom Proteom, 2016, 20: 151-158. DOI:10.1016/j.cbd.2016.10.001 |