 2020, Vol. 31
2020, Vol. 31扩展功能
文章信息
- 余竹梅, 熊衍文, 王文, 张永振
- YU Zhu-mei, XIONG Yan-wen, WANG Wen, ZHANG Yong-zhen
- 中国北方地区蜱携带荆门蜱病毒的分子流行特征分析
- Analysis of molecular epidemic characteristics of Jingmen tick virus among ticks in northern China
- 中国媒介生物学及控制杂志, 2020, 31(3): 272-276
- Chin J Vector Biol & Control, 2020, 31(3): 272-276
- 10.11853/j.issn.1003.8280.2020.03.006
-
文章历史
- 收稿日期: 2020-03-30




蜱是重要的人类和动物病原体媒介, 通过叮咬的方式传播病毒。目前, 全球大约有900个蜱种, 分为3个科:隐喙蜱科(Argasidae)、硬蜱科(Ixodidae)和纳蜱科(Nuttalliellidae)[1-2]。到目前为止, 至少有38种病毒通过蜱叮咬传播, 主要包括发热伴血小板减少综合征病毒、克里米亚刚果出血热病毒、非洲猪瘟病毒、森林脑炎病毒、腹地病毒、鹿蜱病毒以及内罗毕绵羊病毒等[1-3], 它们中有的引起人类和动物严重疾病甚至致死。近年来, 基于宏转录组学进一步揭示了蜱传病毒的多样性, 如弹状病毒科、呼肠孤病毒科、黄病毒科和布尼亚病毒目、单负链RNA病毒目的新病毒种发现[2-3]。这些新发现未分类的病毒可能作为共生体存在, 也可能成为重要的病原体影响人类和动物健康。
荆门蜱病毒(Jingmen tick virus, JMTV)是一类单股正链的带包膜RNA病毒, 其基因组由4个节段组成, 其中节段1(S1)和节段3(S3)分别编码非结构蛋白NSP1和NSP2, 节段2(S2)编码结构蛋白VP1, 节段4(S4)分别编码结构蛋白VP2和VP3, 非结构蛋白NSP1和NSP2分别与已知的黄病毒NS5和NS3具有同源性, 而结构蛋白VP1、VP2和VP3尚未发现与已知的病毒同源序列[4]。JMTV首次在湖北省荆门市的蜱中被发现因而命名为JMTV[4]。2016年Ladner等[5]在非洲乌干达的红疣猴又发现1株JMTV(RC27), 在灵长类中的首次发现提示JMTV对人类健康具有潜在威胁。随后与之相似的病毒在巴西的蜱和牛中被再次报道[2, 6]。2018年, 德国和科索沃的研究团队在克里米亚刚果出血热病人血清再次检测到JMTV, 该病毒和人类的关系得到进一步的验证[7]。更为重要的是, Jia等[8]2019年研究证实了在有蜱叮咬史的病人头皮组织、血清中检测到JMTV, 同时他们也在蜱细胞中对该病毒进行了分离培养, 获得了该病毒株。同年, Wang等[9]再次在中国东北地区有蜱叮咬史的发热病人血清中检测并分离到类JMTV样病毒, 命名为阿龙山病毒, 该病毒的发现进一步提示JMTV是一种新的人类蜱传病原体。至此, JMTV得到了更广泛的关注和研究, 病毒分布遍及世界各地, 包括亚洲、非洲、南美洲、加勒比地区和欧洲等[2, 4-13]。
为了进一步了解JMTV在中国北方地区蜱中的流行情况, 我们在中国西北地区的新疆维吾尔自治区(新疆)博乐市、东北的辽宁省沈阳市和北京市昌平区3个地区进行蜱样本采集, 通过反转录-聚合酶链式反应(RT-PCR)方法, 进行了蜱中JMTV分子流行调查。
1 材料与方法 1.1 动物样本采集和处理在新疆博乐市、辽宁省沈阳市和北京市昌平区采集蜱标本, 经形态学鉴定后, 置-80 ℃冰箱保存或进行以下处理:每只蜱分别用磷酸盐缓冲液(PBS)清洗3次, 置于研钵中, 加入400 μl PBS, 充分研磨(一蜱一套研钵器, 遵循一用一消毒原则, 严格无菌操作)[14]。收集研磨液于2 ml的冻存管里, -80 ℃冰箱保存。
1.2 样本总RNA提取, RT-PCR扩增与测序取200 μl蜱悬液, 参照美国Invitrogen公司TRIzol LS Reagent和E.Z.N.A.RNA isolation Kit(OMEGA)说明书提取RNA。使用one step RT-PCR Kit(TaKaRa, 中国大连)试剂盒进行样本RNA中JMTV和类JMTV节段1保守区(S1, 主要编码RdRp)的PCR扩增。One step RT-PCR Kit扩增条件为:50 ℃ 30 min反转录总RNA为cDNA;94 ℃预变性3 min, 94 ℃变性35 s, 53 ℃退火35 s, 72 ℃延伸30 s, 共35个循环;72 ℃10 min。以第1轮产物为模板使用Taq DNA聚合酶(TaKaRa, 中国大连)进行第2轮巢式PCR扩增。Taq DNA聚合酶扩增条件:94 ℃预变性3 min;94 ℃变性35 s, 53 ℃退火35 s, 72 ℃延伸30 s, 共35个循环;72 ℃10 min。筛查引物参照文献[4](表 1)。根据初筛结果进一步进行JMTV全基因组扩增。PCR产物用QIAquick Gel Extraction Kit (Qiagen, 美国)纯化后送生工生物工程(上海)有限公司采用标准Sanger测序法直接测序, 所有测序数据使用Seqman进行组装。

利用Megalign (DNAStar version 5.01)软件进行两两比对同源性分析;利用MEGA 6.0软件中的Clustal W方法进行序列比对, 采用PhyML3.0软件, 最大似然法(Maximum Likelihood, ML)进行遗传距离分析, 构建系统发生树[15-17]。
2 结果 2.1 RT-PCR筛查JMTV在新疆博乐市、辽宁省沈阳市、北京市昌平区3地共采集蜱标本277只, 包括博乐市残缘璃眼蜱(Hyalomma detritum, Hd)83只, 沈阳市长角血蜱(Haemaphysalis Iongicornis, Hl)98只, 昌平区中华革蜱(Dermacentor sinicus, Ds)96只。使用RT-PCR方法对JMTV的S1节段进行检测, 获得阳性标本67只, 阳性率为24.2%, 其中中华革蜱检测阳性41只, 阳性率高达42.7%, 残缘璃眼蜱检出阳性12只, 阳性率为14.5%, 长角血蜱检出阳性14只, 阳性率为14.3%。病毒序列经美国国立生物技术信息中心(NCBI)数据库进行BLASTN比对分析, 新发现的蜱传JMTV与中国其他地区发现的JMTV同源性最高, 核苷酸同源性为91.0%~99.0%, 呈现出病毒遗传多样性。同时我们也进行了阿龙山病毒和分颗粒Guaico库蚊病毒(Guaico Culex virus, GCXV)的检测, 均无阳性检出。
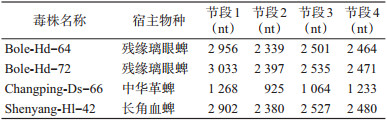
2.2 同源性分析根据初筛阳性样本, 通过RT-PCR扩增获得了4株接近全长的JMTV基因组序列, 根据病毒的宿主和采样地点将这4株毒株分别命名为Bole-Hd-64、Bole-Hd-72、Changping-Ds-66和Shenyang-Hl-42(表 2), 其中前2株来自新疆的残缘璃眼蜱, 后2株分别来自昌平区的中华革蜱和沈阳市的长角血蜱。利用MegAlign软件进行序列比对同源性分析, 显示新发现JMTV4个片段之间核苷酸同源性为91.4%~99.8%, 进一步分析可以看到, 新扩增的JMTV在节段1、节段2和节段4都与已知的新疆啮齿类来源的JMTV同源性最高, 达94.8%~99.8%, 与来自中国其他地区的蜱源性JMTV同源性为90.1%~94.1%。然而节段3中的其中2株病毒(Bole-Hd-72和Shenyang-Hl-42)和新疆啮齿类JMTV同源性高达98.9%~99.1%, 另2株(Bole-Hd-64和Changping-Ds-66)与中国其他地区的蜱源性JMTV同源性为91.6%~93.6%;新扩增的蜱JMTV与来自非洲、南美洲和欧洲的JMTV同源性为71.8%~92.3%, 而与阿龙山病毒同源性相对较低(57.9%~74.2%)。
|
使用PhyML 3.0软件, 以ML构建进化树。由图 1、2可以看出, 4个进化树都分成了3个进化群, 第一进化群由来自亚洲、非洲和南美洲的JMTV组成, 第二进化群由来自科索沃、安纳托利亚和多巴哥的JMTV组成, 最外围的进化群由来自中国、芬兰和法国的阿龙山病毒组成。进一步分析, 在第一进化群中所有JMTV聚集形成了3个明显的分支(图 1、2), 包括中国和老挝JMTV, 乌干达JMTV, 巴西和几内亚JMTV。第Ⅰ亚支, 所有中国来源的JMTV与1株老挝的蜱JMTV聚集到一起, 呈现一定的病毒多样性, 其中在S1、S2、S4节段, 4个新发现的蜱JMTV与新疆啮齿类JMTV聚集在一起, 然而在S3节段, Bole-Hd-72和Shenyang-Hl-42与新疆啮齿类JMTV聚集在一起, 但Bole-Hd-64和Changping- Ds-66与中国其他地区的JMTV聚集在一起, 表明在S3节段JMTV可能发生了基因重排(图 1、2)。

|
| 图 1 基于荆门蜱病毒节段1和节段3核苷酸序列构建的系统发生树 Figure 1 Phylogenetic tree of JMTV based on the nucleotide sequences of segments 1 and 3 |
| |

|
| 图 2 基于荆门蜱病毒节段2和节段4核苷酸序列构建的系统发生树 Figure 2 Phylogenetic tree of JMTV based on the nucleotide sequences of segments 2 and 4 |
| |
JMTV 2010年首次在湖北省荆门市发现, 随着研究的不断深入, 近年来JMTV陆续在亚洲、非洲、欧洲、南美洲和加勒比地区等全球范围内多个国家被报道[2, 4-13]。JMTV的宿主包括蜱、蚊等节肢动物[2, 4, 6, 10-13], 能感染多种哺乳动物如啮齿类、牛、猴、人等[5, 7-9]。本研究通过RT-PCR扩增获得4个新的蜱传JMTV, 分别命名为Bole-Hd-64、Bole-Hd-72、Changping-Ds-66和Shenyang-Hl-42。同源性分析显示, 新发现的蜱JMTV与已知的中国其他地区JMTV同源性最高(90.1%~99.8%), 呈现出病毒遗传多样性。系统发育树显示, 在S3节段4株病毒形成2个独立的进化支, 与中国其他地区的核苷酸差异达9.0%, Bole-Hd-64和Changping-Ds-66基因组在S3节段可能存在基因重排(图 1)。对于这一现象, Qin等[4]2014年首次进行了分析研究, 基因重排也发生在S3节段上(JMTV85/R. microplus/Jingmen/China), 该毒株基因组序列来自湖北省荆门市的微小扇头蜱(Rhipicephalus microplus)[4], 重排现象的发生使得JMTV基因组结构变得更加复杂多样。
蜱作为JMTV的传播媒介, 在该病毒从节肢动物到哺乳动物的传播中扮演着重要角色。4个进化树中, 每一个亚支都包含有来自蜱和来自哺乳动物的JMTV, 而且蜱具有多样性, 包括微小扇头蜱、残缘璃眼蜱、长角血蜱、中华革蜱、爪哇花蜱(Amblyomma javanense)、龟形花蜱(A. testudinarium)、囊型扇头蜱(Rh. bursa)、血红扇头蜱(Rh. sanguineus)、嘉氏扇头蜱(Rh. geigyi)等, 充分地证明了JMTV是以蜱作为传播媒介的虫媒病毒[2, 4, 6, 10-13]。本次研究我们在中国的西北地区新疆博乐市、东北辽宁省沈阳市和北京市昌平区3个地区3个蜱种中进行JMTV的检测工作, 其中长角血蜱携带JMTV已有报道〔4〕, 而中华革蜱和残缘璃眼蜱为首次检测到该病毒, 3个蜱种的阳性率中华革蜱(42.7%)远高于长角血蜱(14.3%)和残缘璃眼蜱(14.5%), 提示中华革蜱可能为JMTV传播的又一优势种群。
系统发生树显示, 新发现的2株毒株(Bole-Hd-72和Shenyang-Hl-42)在4个基因组节段上都与新疆啮齿类JMTV聚集在一起, 同源性高达98.9%~99.1%, 提示在节肢动物和哺乳动物之间病毒可能发生快速的水平传播, 这一理论还有待进一步的研究。啮齿类作为最大类群的哺乳动物, 携带多种病原体, 与人类的关系密切, 同时它也是蜱的寄生宿主[18-25]。因此, 新发现的蜱JMTV与啮齿类JMTV的高度同源性更提示了该病毒对动物和人的感染潜力以及可能的致病性。
2019年, Jia等[8]和Wang等[9]先后报道在有蜱叮咬史的发热病人中检测出JMTV和类JMTV, 表明JMTV对人的致病性以及重要的公共卫生学意义, JMTV因此受到越来越多的关注和研究。鉴于目前研究的局限性, 未来进一步扩大研究的宿主范围和地理分布, 探索JMTV对动物和人的致病性, 感染人的机制, 以及JMTV在生物学上特殊的进化机制, 将有着重要的意义。
| [1] |
Mansfield KL, Lv JZ, Phipps LP, et al. Emerging tick-borne viruses in the twenty-first century[J]. Front Cell Infect Microbiol, 2017, 7: 298. DOI:10.3389/fcimb.2017.00298 |
| [2] |
De Souza WM, Fumagalli MJ, de Oliveira Torres Carrasco A, et al. Viral diversity of Rhipicephalus microplus parasitizing cattle in southern Brazil[J]. Sci Rep, 2018, 8(1): 16315. DOI:10.1038/s41598-018-34630-1 |
| [3] |
Zhang YZ, He YW, Dai YA, et al. Hemorrhagic fever caused by a novel Bunyavirus in China:pathogenesis and correlates of fatal outcome[J]. Clin Infect Dis, 2012, 54(4): 527-533. DOI:10.1093/cid/cir804 |
| [4] |
Qin XC, Shi M, Tian JH, et al. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors[J]. Proc Natl Acad Sci USA, 2014, 111(18): 6744-6749. DOI:10.1073/pnas.1324194111 |
| [5] |
Ladner JT, Wiley MR, Beitzel B, et al. A multicomponent animal virus isolated from mosquitoes[J]. Cell Host Microbe, 2016, 20(3): 357-367. DOI:10.1016/j.chom.2016.07.011 |
| [6] |
Villa EC, Maruyama SR, de Miranda-Santos IKF, et al. Complete coding genome sequence for Mogiana tick virus, a Jingmenvirus isolated from ticks in Brazil[J]. Genome Announc, 2017, 5(18): e00232-17. DOI:10.1128/genomeA.00232-17 |
| [7] |
Emmerich P, Jakupi X, von Possel R, et al. Viral metagenomics, genetic and evolutionary characteristics of Crimean-Congo hemorrhagic fever Orthonairovirus in humans, Kosovo[J]. Infect Genet Evol, 2018, 65: 6-11. DOI:10.1016/j.meegid.2018.07.010 |
| [8] |
Jia N, Liu HB, Ni XB, et al. Emergence of human infection with Jingmen tick virus in China:a retrospective study[J]. EBioMedicine, 2019, 43: 317-324. DOI:10.1016/j.ebiom.2019.04.004 |
| [9] |
Wang ZD, Wang B, Wei F, et al. A new segmented virus associated with human febrile illness in China[J]. N Engl J Med, 2019, 380(22): 2116-2125. DOI:10.1056/NEJMoa1805068 |
| [10] |
Sameroff S, Tokarz R, Charles RA, et al. Viral diversity of tick species parasitizing cattle and dogs in Trinidad and Tobago[J]. Sci Rep, 2019, 9(1): 10421. DOI:10.1038/s41598-019-46914-1 |
| [11] |
Kuivanen S, Levanov L, Kareinen L, et al. Detection of novel tick-borne pathogen, Alongshan virus, in Ixodes ricinus ticks, south-eastern Finland, 2019[J]. Eurosurveillance, 2019, 24(27): 1900394. DOI:10.2807/1560-7917.ES.2019.24.27.1900394 |
| [12] |
Temmam S, Bigot T, Chrétien D, et al. Insights into the host range, genetic diversity, and geographical distribution of Jingmenviruses[J]. mSphere, 2019, 4(6): e00645-19. DOI:10.1128/mSphere.00645-19 |
| [13] |
Dinçer E, Hacıoǧlu S, Kar S, et al. Survey and characterization of Jingmen tick virus variants[J]. Viruses, 2019, 11(11): 1071. DOI:10.3390/v11111071 |
| [14] |
宋凯丽, 熊衍文, 刁秀念, 等. 新疆维吾尔自治区蜱携带白蛉病毒属病毒遗传特征分析[J]. 疾病监测, 2018, 33(8): 670-673. Song KL, Xiong YW, Diao XN, et al. Genetic characteristics of Phlebovirus carried by ticks in Xinjiang Uygur autonomous region[J]. Dis Surveill, 2018, 33(8): 670-673. DOI:10.3784/j.issn.1003-9961.2018.08.014 |
| [15] |
Tamura K, Stecher G, Peterson D, et al. MEGA6:molecular evolutionary genetics analysis version 6.0[J]. Mol Biol Evol, 2013, 30(12): 2725-2729. DOI:10.1093/molbev/mst197 |
| [16] |
Guindon S, Dufayard JF, Lefort V, et al. New algorithms and methods to estimate maximum-likelihood phylogenies:assessing the performance of PhyML 3.0[J]. Syst Biol, 2010, 59(3): 307-321. DOI:10.1093/sysbio/syq010 |
| [17] |
Posada D. jModelTest:phylogenetic model averaging[J]. Mol Biol Evol, 2008, 25(7): 1253-1256. DOI:10.1093/molbev/msn083 |
| [18] |
Guo WP, Lin XD, Wang W, et al. Phylogeny and origins of hantaviruses harbored by bats, insectivores, and rodents[J]. PLoS Pathog, 2013, 9(2): e1003159. DOI:10.1371/journal.ppat.1003159 |
| [19] |
Li K, Lin XD, Wang W, et al. Isolation and characterization of a novel arenavirus harbored by rodents and shrews in Zhejiang province, China[J]. Virology, 2015, 476: 37-42. DOI:10.1016/j.virol.2014.11.026 |
| [20] |
Li K, Lin XD, Huang KY, et al. Identification of novel and diverse rotaviruses in rodents and insectivores, and evidence of cross-species transmission into humans[J]. Virology, 2016, 494: 168-177. DOI:10.1016/j.virol.2016.04.017 |
| [21] |
Meerburg BG, Singleton GR, Kijlstra A. Rodent-borne diseases and their risks for public health[J]. Crit Rev Microbiol, 2009, 35(3): 221-270. DOI:10.1080/10408410902989837 |
| [22] |
Wang W, Lin XD, Guo WP, et al. Discovery, diversity and evolution of novel coronaviruses sampled from rodents in China[J]. Virology, 2015, 474: 19-27. DOI:10.1016/j.virol.2014.10.017 |
| [23] |
Oldal M, Sironen T, Henttonen H, et al. Serologic survey of orthopoxvirus infection among rodents in Hungary[J]. Vector Borne Zoonotic Dis, 2015, 15(5): 317-322. DOI:10.1089/vbz.2014.1731 |
| [24] |
Berto A, Anh PH, Carrique-Mas JJ, et al. Detection of potentially novel paramyxovirus and coronavirus viral RNA in bats and rats in the Mekong Delta region of southern Viet Nam[J]. Zoonoses Public Health, 2018, 65(1): 30-42. DOI:10.1111/zph.12362 |
| [25] |
Chen JT, Qin J, Li K, et al. Identification and characterization of a novel subtype of Tula virus in Microtus arvalis obscurus voles sampled from Xinjiang, China[J]. Infect Genet Evol, 2019, 75: 104012. DOI:10.1016/j.meegid.2019.104012 |