 2019, Vol. 30
2019, Vol. 30扩展功能
文章信息
- 李思思, 张晓雨, 张艳凯, 刘敬泽
- LI Si-si, ZHANG Xiao-yu, ZHANG Yan-kai, LIU Jing-ze
- 内蒙古地区全沟硬蜱细菌群落结构及多样性分析
- An analysis of bacterial community composition and diversity in Ixodes persulcatus in Inner Mongolia, China
- 中国媒介生物学及控制杂志, 2019, 30(6): 607-612
- Chin J Vector Biol & Control, 2019, 30(6): 607-612
- 10.11853/j.issn.1003.8280.2019.06.003
-
文章历史
- 收稿日期: 2019-08-05
- 网络出版时间: 2019-10-15 09:46




2 衡水学院生命科学学院, 河北 衡水 053000
2 College of Life Sciences, Hengshui University
蜱是一类专性吸血的体外寄生虫,在世界范围内分布广泛。蜱通常寄生于家畜、野生动物和鸟类体表,也常侵袭人类,是引起人、畜多种感染性和寄生性疾病的传播媒介。由蜱传播的病原体包括螺旋体、立克次体(Rickettsia)、巴贝斯虫、巴尔通体、考克次体(Coxiella)、土拉弗朗西斯菌(土拉菌)及病毒等,引起莱姆病、肾综合征出血热、人粒细胞无形体病、非洲猪瘟等自然疫源性人畜传染病[1],给人类健康、畜牧业发展和野生动物保护带来极大危害。
全沟硬蜱(Ixodes persulcatus)属于硬蜱科(Ixodidae)硬蜱属(Ixodes),是北方针阔混交林带的优势种,主要分布在波罗的海到俄罗斯远东地区。在我国新疆维吾尔自治区、内蒙古自治区、东北3省、河北省北部等地区均为优势蜱种[2-4]。它是典型的3宿主蜱,完成一个世代需经过卵、幼蜱、若蜱和成蜱4个阶段。自卵孵出后,全沟硬蜱通常需要更换多个宿主完成饱血过程,这使得蜱体内携带的细菌种类增多。病原体的多重感染在全沟硬蜱中普遍存在。Eremeeva等[2]在全沟硬蜱中同时检测到伯氏疏螺旋体(Borrelia burgdorferi)、埃立克体(Ehrlichia)、立克次体。国内学者针对全沟硬蜱携菌研究,也发现2种或以上细菌共感染的情况[4-6]。
高通量测序技术能够完成基因组的高覆盖率测序,且价格低廉,分析基因组的各项数据也更加全面深入。高通量测序技术在蜱类细菌群落分析中发挥着重要作用。Andreotti等[7]首次应用高通量测序技术在微小扇头蜱〔Rhipicephalus(Boophilus)microplus〕中检测到包括沃尔巴克氏体(Wolbachia)、考克次体、疏螺旋体(Borrelia)在内的共计121个属的细菌。之后,应用高通量技术在多种蜱中发现了更多新的共生菌及病原体,以及在蜱中肠、卵巢、马氏管等组织中特异存在的细菌种类[7-9]。细菌多样性与宿主蜱的生物学及其携带、传播病原体的能力密切相关[8, 10],截至到目前,关于全沟硬蜱的研究多集中于病原体的检测,对其细菌多样性和共生菌的研究相对较少。因此,应用高通量技术对蜱内细菌多样性展开研究,能够揭示宿主与体内微生物、微生物群落之间复杂的生态联系,并为蜱的生物防控提供新的思路和途径。
1 材料与方法 1.1 样本采集全沟硬蜱样品采集自内蒙古额尔古纳国家级自然保护区(120°58′02″E,51°29′25″N,图 1)。自然保护区位于大兴安岭西北坡,地处中国与俄罗斯边境,与俄罗斯只有一江之隔。山地是保护区地貌的主体。保护区冬季最低温度可达-44.5 ℃,夏季最高温度为29.8 ℃,年平均气温为6.4 ℃。属于大兴安岭山地寒温带湿润林业气候区,并具有大陆性季风气候特征。野外采用拖旗法采集自由生活的全沟硬蜱饥饿成蜱,雌蜱(Ip-F)、雄蜱(Ip-M)、卵(Ip-E,来自野外饱血雌蜱)立刻浸泡在95%乙醇溶液中,带回实验室置于-80 ℃,用于DNA提取和PCR扩增。

|
注: 表示样品采集地
图 1 全沟硬蜱在我国的地理分布(图中 表示样品采集地
图 1 全沟硬蜱在我国的地理分布(图中  |
| |
取出在95%乙醇溶液中浸泡的全沟硬蜱饥饿成蜱,雌、雄蜱各10只,卵100只,分别用75%乙醇溶液进行3次漂洗,以去除表面污染,然后在无菌水中用移液器吹洗去除体表乙醇[11]。使用微波炉高温灭菌后的研磨棒在1.5 ml EP管中对样品进行研磨,然后依照DNA提取试剂盒(QIAGEN,美国)的操作步骤进行总DNA的提取。
1.3 细菌16S rDNA基因V3区高通量测序 1.3.1 引物合成与PCR扩增收集纯化基因组DNA,16SrDNA基因通用引物PCR扩增。按指定测序区域,合成带有5′ 454A、B接头-特异引物3′的融合引物,细菌核糖体16S rDNA基因保守区域中包括了V1、V2、V3和V4 4个高变区,PCR扩增引物为V3保守区通用引物(表 1),A为测序端,在通用引物外侧加入标签Tag序列以区分数据,B端引物可共用。扩增产物纯化后,连接A和B接头,Adaptor A(5′GCCTCCCTCGCGCCATCAG-3′)和Adaptor B(5′GCCTTGCCAGCCCGCTCAG-3′),使用磁珠筛选去除接头自连片段;片段筛选,保留一端是A接头、一端是B接头的片段;氢氧化钠变性,产生单链DNA片段。

参照电泳初步定量结果,将PCR产物用QuantiFluorTM-ST蓝色荧光定量系统(Promega,USA)进行检测定量,之后按照每个样品的测序量要求,进行相应比例的混合。
1.3.3 高通量测序依靠Roche Genome Sequencer FLX测序平台(Shanghai Majorbio Biopharm Technology Co.,Ltd,China上海美吉生物医药科技有限公司)完成上机检测,读取序列信息,从而得到测序原始序列(Rawreads)。
1.4 操作分类单元(OTU)聚类与分类学分析利用mothur软件(http://www.mothur.org/wiki/Classify.seqs)将Tags进行分类单元OTU聚类、分析和物种注释,通过计算多样性指数和绘制稀释曲线进行样本复杂度分析(α-多样性),算法为Na?ve Bayesian Classifier[12]。
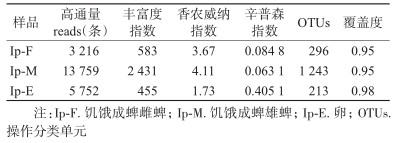
2 结果 2.1 全沟硬蜱16S rDNA基因测序结果及物种复杂度分析在全沟硬蜱雌、雄、卵3个样本中共获得22 727条序列,以雄蜱所含最多,为13 759条,其次分别为卵和雌蜱。所有样本的覆盖度均≥0.95,表明本次测序深度基本可覆盖所有细菌物种。其中,雄蜱样本的丰富度指数(Chao 1)、多样性指数最高,其次为雌蜱和卵。香农威纳指数(Shannon index)、辛普森指数(Simpson index)等见表 2。
|
所有序列通过聚类分析,共生成1 570个OTUs,在雌、雄、卵3个样品中分别为296、1 243和213个,其中有31个OTU在3个样品中均有出现,样本中单独出现的OTU以雄蜱最多为1 127个,雌蜱为212个,卵为120个(图 2)。

|
| 注:Ip-F.饥饿成蜱雌蜱;Ip-M.饥饿成蜱雄蜱;Ip-E.卵 图 2 全沟硬蜱不同样品中的OTUs聚类分布 Figure 2 Distribution of clustered operational taxonomic units in different Ixodes persulcatus samples |
| |
在门水平上,3个样本携带的相关细菌隶属于20个门,每个样本中均出现未被鉴定的门类(图 3A)。此外,所含门类最多的为雄蜱(16门),其次为雌蜱(9门)和卵(9门)。变形菌门(Proteobacteria)为3个样品共有的优势菌门,其在卵中相对丰度最高,约为96.97%,其次为雄蜱,约为61.92%,雌蜱最小,约为26.55%。其他共有的菌门包括厚壁菌门(Firmicutes)、放线菌门(Actinobacteria)和栖热菌门(Deinococcus- Thermus)等。其中,厚壁菌门在雌蜱与卵中相对丰度较高,放线菌门在雌蜱中相对丰度较高。

|
| 注:Ip-F.饥饿成蜱雌蜱;Ip-M.饥饿成蜱雄蜱;Ip-E.卵 图 3 全沟硬蜱微生物群落结构 Figure 3 Bacterial community composition in Ixodes persulcatus |
| |
在纲水平上,3个样本均携带相对丰度较高的菌纲依次包括α-变形菌纲(Alphaproteobacteria)、杆菌纲(Bacilli)、γ-变形菌纲(Gammaproteobacteria)、放线菌纲(Actinobacteria)、异常球菌纲(Deinococci)和β-变形菌纲(Betaproteobacteria),每个样本中均出现未被鉴定的菌纲(图 3B)。所含菌纲数最多的为雄蜱(26纲),其次为雌蜱(15纲)和卵(13纲)。
在属水平上,不同样本中相对丰度较高的菌属组成差异较大,雌蜱中居前5位的是葡萄球菌属(Staphylococcus,40.36%)、枯草芽孢属(Brevibacterium,19.09%)、立克次体属(Rickettsia,17.60%)、假丝酵母属(Candidatus anadelfobacter,5.22%)和短状杆菌属(Brachybacterium,3.30%),雄蜱中居前5位的是弧菌属(Vibrio,38.67%)、明串珠菌属(Trichococcus,36.85%)、假丝酵母属(12.52%)、立克次体属(3.87%)和假交替单胞菌属(Pseudoalteromonas,2.36%),卵中居前3位的是假丝酵母属(72.98%)、立克次体属(22.10%)和栖热菌属(Thermus,2.50%)(图 3C)。
在所有检测到的菌属中,含部分共生菌,如Can. Montezuma、拟杆菌(Bacteroides)、肠杆菌(Enterobacter)等;以及病原体与条件致病菌,如3个样本中均检测到的塔拉塞维奇立克次体(Can. Rickettsia tarasevichiae)和疏螺旋体等;此外还有大量环境细菌(表 3)。

与其他节肢动物一样,蜱也拥有自己独特的细菌群落,这些细菌群落与蜱宿主具有互利共生或寄生关系[7]。本研究利用罗氏454测序技术对采自内蒙古的全沟硬蜱细菌群落进行了分析。在雌蜱、雄蜱和卵3个样本中,共检测到细菌20门,其中变形菌门、厚壁菌门和放线菌门为优势菌。这一结果与我国黑龙江省牡丹江市、俄罗斯Novosibirsk地区等报告的全沟硬蜱细菌群落组成一致[3-4, 14]。同时与其他蜱种,如长角血蜱(Haemaphysalis longicornis)[15]、褐黄血蜱(H. flava)[16]、西藏血蜱(H. tibetensis)[17]和日本锐蜱(Argas japanicus)[11]的研究结果类似。表明蜱类细菌组成在门分类阶元,主要以变形菌门为优势菌,且丰度较高的菌门种类组成相近,但各个菌门所占比例在不同蜱种间存在一定差异。
全沟硬蜱中共鉴定出细菌107个属,以病原体居多。群落多样性分析表明不同样本间携带的细菌种类和相对丰度差异较大,以雄蜱所检出的菌种最多,可能因找寻宿主吸血及寻觅配偶使其活动范围增加,同时也与雌雄体不同生理机制有关。蜱中较丰富的菌属包括葡萄球菌属、弧菌属、明串珠菌属、假丝酵母属和立克次体属等。3个样本中均检测到斑点热群立克次体(spotted fever group rickettsiae,SFGR)。在我国的蜱类中至少检测到8种经过验证的SFGR。硬蜱科包括革蜱属、硬蜱属、扇头蜱属、血蜱属、璃眼蜱属和花蜱属均确定感染立克次体[18-19]。SFGR最初发现于俄罗斯,包括乌拉尔南部和西伯利亚西部和东部[20],也曾在日本和爱沙尼亚的全沟硬蜱中发现[21-22]。直到2013年中国东北地区的5名患者被诊断出该病原体时,SFGR才被认为是致病性的[23-24]。SFGR可以通过蜱叮咬动物,在宿主动物中传播,也可以通过蜱经期传播至下一个生活史阶段,还可经卵传播给子代。
埃立克体为专性细胞内寄生菌,一般实验室难以体外培养,可引起人畜共患疾病。查菲埃立克体(E. chaffeensis)是人单核细胞埃立克体病的病原体,对其研究较为广泛。1999年,首次在俄罗斯全沟硬蜱中发现鼠埃立克体(E. muris)[25]。之后,其在西伯利亚全沟硬蜱中亦被检测到[3]。基于16S rDNA基因寡核苷酸序列分析和同源性比较,曹务春等[26]将埃立克体分为3个不同的基因组,其中查菲埃立克体和鼠埃立克体同属于一组。高东旗等[27]在内蒙古莫尔道嘎采集的全沟硬蜱、森林革蜱(D. silvarum)、嗜群血蜱(H. concinna)以及从新疆精河采集的全沟硬蜱和草原革蜱(D. nutalli)中检测到埃立克体,其与鼠埃立克体亲缘关系接近,核苷酸同源性高达99.4%。之后,刘欢欢[28]首次在吉林和黑龙江省全沟硬蜱检测到鼠埃立克体,阳性率为4.3%~1.9%。2016年,我国首次从人体内发现该病原体的存在[29],表明在欧亚大陆部分国家和地区,全沟硬蜱可能为鼠埃立克体的主要传播媒介,通过叮咬可引起人感染埃里克体病。
疏螺旋体是一种细菌,经蜱传播能引起人类感染回归热,是世界上最常见的媒介传播疾病之一。在全沟硬蜱卵中检测到的宫本疏螺旋体,属于螺旋体属,Takano等[30]在日本北海道地区,发现全沟硬蜱和巴甫洛夫斯克硬蜱(I. pavlovskyi)携带宫本疏螺旋体较高,可能是引起当地人群回归热最重要的传播媒介。宫本疏螺旋体最早发现于俄罗斯,之后在北美、欧洲均有人感染病例出现。Kurilshikov等[3]应用高通量技术,检测到在西伯利亚地区的全沟硬蜱中也有宫本疏螺旋体感染。Mirza[14]发现在全沟硬蜱中出现立克次体与螺旋体共感染的情况。本研究在全沟硬蜱的卵中检测到宫本疏螺旋体,可能是来自父母亲本,表明内蒙古地区全沟硬蜱有传播莱姆病的潜在威胁。
全沟硬蜱中除病原体外,还有大量环境细菌,如金黄杆菌属、欧文菌、不动杆菌、短波单胞菌、志贺埃希菌、白杆菌属、假单胞菌、中间气单胞菌、不动杆菌和沙雷菌等,这些细菌在环境中普遍存在,有一些则是潜在的人畜致病菌,即使某些细菌含量很低,依然值得人们关注。另有丛毛单胞菌、平螺旋菌科、克雷杆菌等,仅在卵中被检测到,这些细菌是否由于样品污染,还有待进一步验证。此外,还发现多种蜱共生菌,包括肠道微生物拟杆菌、肠杆菌,以及母系遗传共生菌,如细菌Montezuma,属于类立克次体胞内共生菌,可经卵传递至子代,其在雄性后代中的感染率逐渐降低或完全消失,曾有研究表明,Montezuma在全沟硬蜱雌蜱中的感染率明显高于雄蜱[18],而本研究显示Montezuma在卵、雄蜱、雌蜱中的相对丰度分别为72.57%、12.41%和5.16%,可能与实验样本大小有关。此外,该菌在巴甫洛夫斯克硬蜱中也曾被发现[3]。
本研究通过高通量测序技术对内蒙古额尔古纳国家级自然保护区全沟硬蜱携带的菌群进行分析,检测到大量病原体与环境致病菌,但未检出土拉菌、考克次体、耶尔森菌(Yersinia)[3-4, 7, 11]。全沟硬蜱携菌的主要原因可能包括:①该蜱在保护区活动范围较广,因与土壤、水源接触而直接感染;②蜱通过吸食宿主血液,因宿主体表或体内含有致病菌,通过寄生带入蜱体内[4];③对于蜱内的共生菌,可能通过母系传递到子代[3, 8]。内蒙古额尔古纳自然保护区位于中俄边境,由于宿主动物迁移、人为活动等原因会使境外蜱越境进入中国,俄罗斯境内蜱传病原体的感染率和复合感染情况远高于中国,因此针对保护区内蜱和蜱传病原体的研究,对我国蜱传疾病的有效监测和预防具有重要意义。
| [1] |
张艳凯, 刘敬泽. 蜱类病原体和共生菌多样性及其作用[J]. 应用昆虫学报, 2019, 56(1): 1-11. DOI:10.7679/j.issn.2095-1353.2019.001 |
| [2] |
Eremeeva ME, Oliveira A, Moriarity J, et al. Detection and identification of bacterial agents in Ixodes persulcatus Schulze ticks from the north western region of Russia[J]. Vector Borne Zoonotic Dis, 2007, 7(3): 426-436. DOI:10.1089/vbz.2007.0112 |
| [3] |
Kurilshikov A, Livanova NN, Fomenko NV, et al. Comparative metagenomic profiling of symbiotic bacterial communities associated with Ixodes persulcatus, Ixodes pavlovskyi and Dermacentor reticulatus ticks[J]. PLoS One, 2015, 10(7): e0131413. DOI:10.1371/journal.pone.0131413 |
| [4] |
Zhang XC, Yang ZN, Lu B, et al. The composition and transmission of microbiome in hard tick, Ixodes persulcatus, during blood meal[J]. Ticks Tick-Borne Dis, 2014, 5(6): 864-870. DOI:10.1016/j.ttbdis.2014.07.009 |
| [5] |
程成.黑龙江口岸蜱媒病原的多样性及其复合感染调查[D].北京: 中国人民解放军军事医学科学院, 2017.
|
| [6] |
潘玉平, 杨吉飞, 牛庆丽, 等. 黑龙江省全沟硬蜱携带伯氏疏螺旋体广义种和斑点热群立克次体的研究[J]. 中国兽医科学, 2017, 47(1): 31-37. DOI:10.16656/j.issn.1673-4696.2017.01.005 |
| [7] |
Andreotti R, de León AAP, Dowd SE, et al. Assessment of bacterial diversity in the cattle tick Rhipicephalus (Boophilus) microplus through tag-encoded pyrosequencing[J]. BMC Microbiol, 2011, 11: 6. DOI:10.1186/1471-2180-11-6 |
| [8] |
Narasimhan S, Fikrig E. Tick microbiome:the force within[J]. Trends Parasitol, 2015, 31(7): 315-323. DOI:10.1016/j.pt.2015.03.010 |
| [9] |
Greay TL, Gofton AW, Paparini A, et al. Recent insights into the tick microbiome gained through next-generation sequencing[J]. Parasite Vector, 2018, 11: 12. DOI:10.1186/s13071-017-2550-5 |
| [10] |
Bonnet SI, Binetruy F, Hernández-Jarguín AM, et al. The tick microbiome:why non-pathogenic microorganisms matter in tick biology and pathogen transmission[J]. Front Cell Infect Microbiol, 2017, 7: 236. DOI:10.3389/fcimb.2017.00236 |
| [11] |
Yan P, Qiu ZX, Zhang TT, et al. Microbial diversity in the tick Argas japonicus (Acari:Argasidae) with a focus on Rickettsia pathogens[J]. Med Vet Entomol, 2019, 33(3): 327-335. DOI:10.1111/mve.12373 |
| [12] |
Wang Q, Garrity GM, Tiedje JM, et al. Naïve Bayesian Classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J]. Appl Environ Microbiol, 2007, 73(16): 5261-5267. DOI:10.1128/AEM.00062-07 |
| [13] |
杨瑞馥, 陶天申, 方呈祥, 等. 细菌名称双解及分类词典[M]. 北京: 化学工业出版社, 2011: 10.
|
| [14] |
Mirza OA.利用反向线状印迹及高通量技术分别检测蜱体内梨形虫及全沟硬蜱体内细菌研究[D].北京: 中国农业科学院, 2016.
|
| [15] |
韩娜, 张琳, 张雯, 等. 长角血蜱细菌群落结构及多样性研究[J]. 中国媒介生物学及控制杂志, 2016, 27(5): 426-431. DOI:10.11853/j.issn.1003.8280.2016.05.002 |
| [16] |
邹安迪.褐黄血蜱的带菌分析[D].长沙: 湖南农业大学, 2016.
|
| [17] |
Yu ZJ, Wang RR, Li NX, et al. Microbial diversity of the Tibetan tick Haemaphysalis tibetensis (Acari:Ixodidae)[J]. Exp Appl Acarol, 2017, 73(2): 237-244. DOI:10.1007/s10493-017-0179-x |
| [18] |
Maina AN, Jiang J, Omulo SA, et al. High prevalence of Rickettsia africae variants in Amblyomma variegatum ticks from domestic mammals in rural western Kenya:implications for human health[J]. Vector Borne Zoonot, 2014, 14(10): 693-702. DOI:10.1089/vbz.2014.1578 |
| [19] |
Lopez-Velez R, Palomar AM, Oteo JA, et al. Novel Candidatus Rickettsia species detected in Nostril tick from human, Gabon, 2014[J]. Emerg Infect Dis, 2015, 21(2): 325-327. DOI:10.3201/eid2102.141048 |
| [20] |
Shpynov S, Fournier PE, Rudakov N, et al. 'Candidatus Rickettsia tarasevichiae' in Ixodes persulcatus ticks collected in Russia[J]. Ann N Y Acad Sci, 2003, 990(1): 162-172. DOI:10.1111/j.1749-6632.2003.tb07358.x |
| [21] |
Inokuma H, Ohashi M, Jilintai, et al. Prevalence of tick-borne Rickettsia and Ehrlichia in Ixodes persulcatus and Ixodes ovatus in Tokachi district, Eastern Hokkaido, Japan[J]. J Vet Med Sci, 2007, 69(6): 661-664. DOI:10.1292/jvms.69.661 |
| [22] |
Katargina O, Geller J, Ivanova A, et al. Detection and identification of Rickettsia species in Ixodes tick populations from Estonia[J]. Ticks Tick-Borne Dis, 2015, 6(6): 689-694. DOI:10.1016/j.ttbdis.2015.06.001 |
| [23] |
Jia N, Zheng YC, Jiang JF, et al. Human infection with Candidatus Rickettsia tarasevichiae[J]. N Engl J Med, 2013, 369(12): 1178-1180. DOI:10.1056/NEJMc1303004 |
| [24] |
Fang LQ, Liu K, Li XL, et al. Emerging tick-borne infections in mainland China:an increasing public health threat[J]. Lancet Infect Dis, 2015, 15(12): 1467-1479. DOI:10.1016/S1473-3099(15)00177-2 |
| [25] |
Ravyn MD, Korenberg EI, Oeding JA, et al. Monocytic Ehrlichia in Ixodes persulcatus ticks from Perm, Russia[J]. Lancet, 1999, 353(9154): 722-723. DOI:10.1016/S0140-6736(98)05640-2 |
| [26] |
曹务春, 张泮河, 张习坦, 等. PCR检测蜱中查菲埃立克体DNA及其序列分析[J]. 寄生虫与医学昆虫学报, 1999, 6(1): 58-63. |
| [27] |
高东旗, 曹务春, 张习坦, 等. 应用半巢式PCR检测我国北方一些蜱种中的查菲埃立克体[J]. 中国媒介生物学及控制杂志, 2000, 11(3): 220-224. DOI:10.3969/j.issn.1003-4692.2000.03.018 |
| [28] |
刘欢欢.吉林省和黑龙江省蜱种分布及3种重要蜱传微生物分子流行病学调查[D].长春: 吉林农业大学, 2017.
|
| [29] |
Yu PF, Niu QL, Liu ZJ, et al. Molecular epidemiological surveillance to assess emergence and re-emergence of tick-borne infections in tick samples from China evaluated by nested PCRs[J]. Acta Trop, 2016, 158: 181-188. DOI:10.1016/j.actatropica.2016.02.027 |
| [30] |
Takano A, Toyomane K, Konnai S, et al. Tick surveillance for relapsing Fever Spirochete Borrelia miyamotoi in Hokkaido, Japan[J]. PLoS One, 2014, 9(8): e104532. DOI:10.1371/journal.pone.0104532 |