 2017, Vol. 28
2017, Vol. 28扩展功能
文章信息
- 陈健, 岳巧云, 邱德义, 刘德星
- CHEN Jian, YUE Qiao-yun, QIU De-yi, LIU De-xing
- 二代测序技术检测大头金蝇携带细菌的研究
- Next Generation Illumina Sequencing of bacteria carried by Chrysomya megacephala
- 中国媒介生物学及控制杂志, 2017, 28(2): 124-130
- Chin J Vector Biol & Control, 2017, 28(2): 124-130
- 10.11853/j.issn.1003.8280.2017.02.007
-
文章历史
- 收稿日期: 2016-12-06
- 网络出版时间: 2017-02-17 09:42




大头金蝇(Chrysomya megacephala)属于双翅目(Diptera)、丽蝇科(Calliphoridae)、金蝇属(Chrysomya),是一种常见蝇类,在全球均有分布。该蝇成虫不仅骚扰人类和家畜,更重要的是很多病原体的传播媒介[1-3]。细菌常规鉴定主要从细菌形态、生理生化或者蛋白质等水平进行[4]。近年来,微生物自动鉴定系统等细菌数值分类法以及利用16S rRNA全序列或者某一段序列进行细菌种类的鉴定方法得到广泛应用[5-7]。细菌的16S rRNA有9个可变区(V1~V9),该可变区序列在不同种的细菌间存在一定差异,可用于细菌种类的鉴定[8-9]。尽管目前已有大量利用微生物自动鉴定系统和16S rRNA的研究报道[10-11],但是这些方法均存在不足,某些自动鉴定系统数据库有限,有时也不能准确地鉴定经过长期培养的细菌种类,仍需要做大量的形态学鉴定、生化反应等补充实验[4, 12-13]。非单一菌株16S rRNA的分子鉴定需扩增转化后挑单克隆进行,或者需要先将细菌进行生化分离培养才可鉴定,极易漏检,耗时长、成本高、通量低[14],而媒介生物携带的细菌不会是单一菌株。自2011年Bartram等[15]开发使用Illumina高通量测序平台研究环境微生物的方法以来,越来越多的研究者开始使用Illumina高通量测序平台检测16S rRNA和16S rRNA不同的可变区,研究环境微生物的物种丰度、微生物多样性或病畜体内病原体的情况[16-18]。
目前关于医学媒介生物携带细菌的研究,大多采用生物化学或传统的分子鉴定方法[2-3]。尚无采用二代测序技术(NGS)研究医学媒介生物携带细菌的报道。本研究首次使用高通量测序平台对大头金蝇体内及其体表携带的细菌进行研究,以此建立一种经济、高效和方便的检测医学媒介生物携带细菌的方法。
1 材料与方法 1.1 样本收集实验用大头金蝇,采用笼诱法捕获。于2013年9月16日,在诱捕笼中放入鱼内脏后置于中山出入境检验检疫局后山花园中进行诱捕。将捕获蝇类置于-20 ℃冰箱冷冻处死并保存。在生物安全柜中使用经75%乙醇擦拭显微镜鉴定其种类,将鉴定后的蝇类置于已灭菌的试管中,于-20 ℃保存备用。
1.2 样本处理 1.2.1 体表携带细菌的收集取3只冷冻处死的大头金蝇置于灭菌的离心管中,加入500 μl 0.9% NaCl溶液,漩涡振荡30 s,离心半径6 cm,8 000 r/min离心3 min,小心吸取上清液450 μl,放入75%乙醇中消毒丢弃,剩下的溶液吹打悬浮后转移到另一只灭菌的离心管中,该过程重复3次。将收集的菌液95 ℃水浴10 min热裂解细菌,离心半径6 cm,12 000 r/min,离心2 min收集上清液,保存在4 ℃冰箱作为模板DNA备用。
1.2.2 体内携带细菌的收集将1.2.1中体表清洗过的3只大头金蝇移入装有1 ml 75%乙醇的离心管中浸泡5 min进行消毒处理,重复3次;然后加入500 μl 0.9% NaCl溶液,漩涡振荡30 s清洗表面残余的乙醇,吸净溶液后将样本放置在生物安全柜中晾干15 min。用灭菌镊子将样本移入新离心管中,将标本研碎成糊状,然后采用体表细菌同样的收集方式收集模板DNA,废弃物置75%乙醇中消毒处理丢弃。
1.3 PCR扩增扩增16S rRNA V3可变区,引物:V3-F:5′-CCA GAC TCC TAC GGG AGG CAG-3′;V3-R:5′-CGT ATT ACC GCG GCT GCT G-3′ [8],引物为Thermo Fisher Scientific Inc广州公司合成。体内外分别扩增。PCR反应体系(100 μl):2×KOD Buffer 50 μl;dNTP(2 mmol/L)5 μl;KOD FX polymerase(Toyobo Life Science Department)2 μ(l 1 U/μl);引物V3-F(20 mmol/L)2 μl;引物V3-R(20 mmol/L)2 μl;模板DNA 6 μl;加ddH2O补足至100 μl。PCR反应条件:94 ℃预变性2 min。98 ℃变性10 s,52 ℃退火30 s,68 ℃延伸30 s,30个循环。68 ℃延伸10 min,10 ℃保温。
1.4 高通量测序和数据分析高通量测序和数据分析参照Capurro等[14]方法。PCR扩增产物送安诺优达基因科技(北京)有限公司测序。若测序公司将混合其他样品在一个反应中进行测序,需PCR产物加上序列标签(Barcodes)进行标记,采用Illumina HiSeq 2000进行测序,测序策略为PE100。原始的测序图谱使用Base Calling of CASAVA软件转化成碱基对,在原始数据中除去含有带接头的、低质量的序列后得到有效序列,再对得到的序列进行引物修饰,最后根据不同标记将所得到的序列进行分组。
运算分类单位(operational taxonomic unit,OTU)的挑选、物种的丰度和多样性分析依据Caporaso等[19]方法进行。将序列相似度>97%的读值归为一类,Alpha多样性分析使用QIIME软件计算[19]。
2 结果 2.1 数据过滤和统计大头金蝇体表细菌序列为884 418条,体内细菌序列为954 088条。修剪完标记、引物、去除低质量的序列后得到有效序列体表为879 144条,体内为947 226条,序列获得率分别为99.40%和99.28%(表 1),说明测序获得的序列质量很高,可进行后续数据分析,且表明样本收集、前处理方法及扩增方法是有效和可行的。

从图 1可以看出,测序质量平均值均超过了Q30,表明测序质量很好。错误率<0.1%的序列,体表为93.91%,体内为94.93%,见表 1。

|
| 注:Q30指测序错误率≤0.1%的碱基数目比例,该指标用于评价整体的测序错误率。 图 1 测序样品质量分布 Figure 1 Sequencing quality distribution based on Qphred value |
| |
物种的丰度和α多样性是用来描述生物多样性的基本指标[20],采用non-phylogeny-based指标研究体内外细菌多样性的差异,采用系统发育多样性——谱系多样性(phylogenetic diversity,PD)-完整树分析(whole tree analysis,WTA)指标分析研究细菌种类[21]。根据V3区扩增出的序列计算得出的α多样性显示,体内和体表所携带的细菌种类数量存在很大差异,但根据不同的指标计算得出的体内外α多样性的变化趋势一致(图 2)。无论是PD值还是检测到的细菌种类数,体表检测的数值均远高于体内的值,表明大头金蝇体表携带细菌的多样性远高于体内。

|
| 注:A.检测到的物种数量(observed species,Sobs),是基于某次实验中观测到的种类数,Sobs越高说明多样性越高;B.谱系多样性(phylogenetic diversity,PD)-完整树分析(whole tree analysis,WTA)是基于进化树进行α多样性分析,PD值越大,说明多样性越高。 图 2 大头金蝇体内外携带细菌的α多样性分析 Figure 2 Alpha diversity of bacteria carried externally and internally |
| |
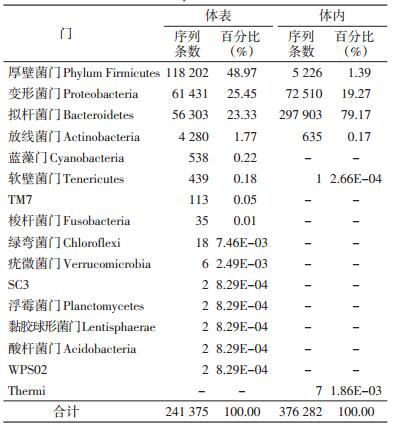
本研究共检测到分属16门的1 492个OTUs,235种可鉴定到属,95种鉴定到种。大头金蝇携带细菌门级分类阶元情况见表 2。大头金蝇体内外携带细菌在门的分类阶元上差异很大,体表携带了15个门的细菌,而体内仅携带了6个门的细菌。但从检测且能确定细菌种类的序列数来看,体内(376 282条)所携带细菌的数量却是体表(241 375条)的1.56倍。无论体内还是体表,厚壁菌门、变形菌门、拟杆菌门的细菌被检测到的概率最大,这3个门的细菌总数在体内外被检测到的概率分别为99.83%和97.75%。其中在体表检测到极少量其他10个门的细菌,Thermi门仅在体内检出。大头金蝇体表携带的细菌种类比体内多,而体内携带的细菌数量较多,体内外所携带的细菌种类存在一定差异。
|
体内外共有95种细菌鉴定到种,大头金蝇体内外携带的细菌情况(种级分类阶元)见图 3。其中,体表携带89种细菌,特有的达54种;体内41种,特有的仅6种;体内外均分布的有35种。

|
| 图 3 大头金蝇体内外携带细菌种数的韦恩分析 Figure 3 Venn analysis of bacterial community at species level carried externally and internally |
| |
由表 3可见,这95种细菌中有40种为致病菌或条件致病菌,这些细菌对人、动物或植物存在不同程度的危害。此外,体表携带细菌中,有5种细菌被检测到的次数超过1 000次,分别为产碱普罗威登斯菌、粪产碱菌、摩氏摩根菌、北纳创联幼虫依格纳季菌和铅黄链球菌。其中产碱普罗威登斯菌、粪产碱菌和摩氏摩根菌是致病菌。在体内携带的细菌中有4种被检测到超过1 000次,均为致病菌,分别是产碱普罗威登斯菌、粪产碱菌、摩氏摩根菌和变形斑病沙雷菌,前3种菌在体表也被发现,仅变形斑病沙雷菌为体内特有。产碱普罗威登斯菌在体内被检测到的次数是体表检测次数的10倍,粪产碱菌在体内被检测到的次数是体表的3倍,说明产碱普罗威登斯菌和粪产碱菌比较适应在大头金蝇体内存活,可以推测其携带和传播这两种细菌的概率较高。

|
将鉴定到种的95种细菌进行了系统发育分析。为便于描述和理解,仅对其中一部分进行详细分析,图 4根据V3高变区,4个属的13个种明确地被聚类为4个不同分支。但是也有例外,Khamis等[22]发现根据16S rRNA序列和rpoB基因建立的系统树,同属的克罗彭施泰特棒状杆菌和变异枝孢菌的亲缘关系并不很近。但研究结果显示,克罗彭施泰特棒状杆菌与所有变异枝孢菌的序列聚在了一起,与Khamis等[22]的研究有一定差异,但符合同属不同种应该聚在一起的理论。

|
| 注:A. 13种细菌的系统进化树;B. 8种细菌的系统进化树。 图 4 基于16S rRNA V3区鉴定到种级水平细菌的系统进化树 Figure 4 Dendrograms of the bacteria at species level based on V3 hypervariable region |
| |
本研究所用的大头金蝇是在中山出入境检验检疫局后山的花园中笼诱获得,该花园经常有居民活动,且临近餐厅和一个垃圾堆放点。大头金蝇携带如此多的细菌,存在传播疾病的极大风险,给人类健康带来较大威胁[22]。本研究测序获得的序列质量很高,可进行后续数据分析。表明样本的收集和前处理方法以及PCR的扩增条件均有效可行,建立的基于Illumina高通量测序平台的检测鉴定新方法有效可行,实现了医学媒介生物携带细菌的经济、高效和高通量检测。该方法省去培养、挑单克隆等操作,细菌损失小,这可能是本研究能发现比前人研究细菌种类更多的原因之一。
本研究旨在探索建立检测鉴定医学媒介生物携带细菌的新方法,选择了可对绝大部分细菌进行分类鉴定且片段较短的V3可变区进行测序[12],而未选择适合鉴定分析更复杂的环境微生物的V4可变区[23],这是给本研究带来的不足之处,因为使用单一的V3可变区不能很好地区分所有细菌种类[11]。也有研究报道,V6区适合鉴定除了肠杆菌外其他所有细菌,V4、V5、V7和V8区不能用于种属鉴定[8]。因此在后续的研究中,将采用V3、V4和V6多可变区组合鉴定医学媒介生物携带的细菌种类。此外,由于样本采集和出于探索方法节约成本的考虑,本研究并未设置实验对照组,后续研究中还需要改进样本的收集处理方法和PCR条件,设置对照组以提高研究的科学严谨性,提高PCR产物的获得率和质量,降低样本数量,实现单一医学媒介生物个体进行检测。
| [1] | Chaiwong T, Sukontason KL, Chaisri U, et al. Effects of human contraceptive on reproduction and offspring in Chrysomya megacephala[J]. Asian Pac J Trop Med, 2011, 4(4) : 259–265 .DOI:10.1016/S1995-7645(11)60082-5. |
| [2] | Sukontason KL, Bunchoo M, Khantawa B, et al. Comparison between Musca domestica and Chrysomya megacephala as carriers of bacteria in northern Thailand[J]. Southeast Asian J Trop Med Public Health, 2007, 38(1) : 38–44 . |
| [3] | 陈拓, 曹晓梅, 邓耀华, 等. 浦东机场口岸蝇类携带病原菌种类及16S rRNA鉴定[J]. 中国媒介生物学及控制杂志, 2012, 23(6): 506–511. |
| [4] | 邓梅葵, 孙迎, 韩雯晴. 细菌鉴定方法[J]. 生物医学工程学进展, 2014, 35(2): 84–88. |
| [5] | Petti CA, Polage CR, Schreckenberger P. The role of 16S rRNA gene sequencing in identification of microorganisms misidentified by conventional methods[J]. J Clin Microbiol, 2005, 43(12) : 6123–6125 .DOI:10.1128/JCM.43.12.6123-6125.2005. |
| [6] | Boutaga K, van Winkelhoff AJ, Vandenbroucke-Grauls CMJE, et al. Periodontal pathogens: a quantitative comparison of anaerobic culture and real-time PCR[J]. FEMS Immunol Med Microbiol, 2005, 45(2) : 191–199 .DOI:10.1016/j.femsim.2005.03.011. |
| [7] | Choi SH, Sung H, Kim SH, et al. Usefulness of a direct 16S rRNA gene PCR assay of percutaneous biopsies or aspirates for etiological diagnosis of vertebral osteomyelitis[J]. Diagn Microbiol Infect Dis, 2014, 78(1) : 75–78 .DOI:10.1016/j.diagmicrobio.2013.10.007. |
| [8] | Chakravorty S, Helb D, Burday M, et al. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria[J]. J Microbiol Methods, 2007, 69(2) : 330–339 .DOI:10.1016/j.mimet.2007.02.005. |
| [9] | Moreau MM, Eades SC, Reinemeyer CR, et al. Illumina sequencing of the V4 hypervariable region 16S rRNA gene reveals extensive changes in bacterial communities in the cecum following carbohydrate oral infusion and development of earlystage acute laminitis in the horse[J]. Vet Microbiol, 2014, 168(2/4) : 436–441 . |
| [10] | Zhang JX, Zhang YB, Quan X, et al. Enhanced anaerobic digestion of organic contaminants containing diverse microbial population by combined microbial electrolysis cell (MEC) and anaerobic reactor under Fe (Ⅲ) reducing conditions[J]. Bioresour Technol, 2013: 273–280 .DOI:10.1016/j.biortech.2013.02.103. |
| [11] | Xiao M, Zhang ZZ, Wang JX, et al. Bacterial community diversity in a low-permeability oil reservoir and its potential for enhancing oil recovery[J]. Bioresour Technol, 2013: 110–116 .DOI:10.1016/j.biortech.2013.08.031. |
| [12] | Becker K, Harmsen D, Mellmann A, et al. Development and evaluation of a quality-controlled ribosomal sequence database for 16S ribosomal DNA based identification of Staphylococcus species[J]. J Clin Microbiol, 2004, 42(11) : 4988–4995 .DOI:10.1128/JCM.42.11.4988-4995.2004. |
| [13] | Bosshard PP, Zbinden R, Abels S, et al. 16S rRNA gene sequencing versus the API 20 NE system and the VITEK 2 IDGNB card for identification of nonfermenting Gram negative bacteria in the clinical laboratory[J]. J Clin Microbiol, 2006, 44(4) : 1359–1366 .DOI:10.1128/JCM.44.4.1359-1366.2006. |
| [14] | Capurro A, Artursson K, Waller KP, et al. Comparison of a commercialized phenotyping system, antimicrobial susceptibility testing, and tuf gene sequence-based genotyping for specieslevel identification of coagulase-negative staphylococci isolated from cases of bovine mastitis[J]. Vet Microbiol, 2009, 134(3/4) : 327–333 . |
| [15] | Bartram AK, Lynch MDJ, Stearns JC, et al. Generation of multimillion sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end illumina reads[J]. Appl Environ Microbiol, 2011, 77(11) : 3846–3852 .DOI:10.1128/AEM.02772-10. |
| [16] | Degnan PH, Ochman H. Illumina-based analysis of microbial community diversity[J]. ISME J, 2012, 6(1) : 183–194 .DOI:10.1038/ismej.2011.74. |
| [17] | Gilbert JA, Meyer F, Antonopoulos D, et al. Meeting report: the terabase metagenomics workshop and the vision of an Earth microbiome project[J]. Stand Genomic Sci, 2009, 3(3) : 243–248 . |
| [18] | Mann E, Schmitz-Esser S, Zebeli Q, et al. Mucosa-associated bacterial microbiome of the gastrointestinal tract of weaned pigs and dynamics linked to dietary calcium-phosphorus[J]. PLoS One, 2014, 9(1) : e86950.DOI:10.1371/journal.pone.0086950. |
| [19] | Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nat Methods, 2010, 7(5) : 335–336 .DOI:10.1038/nmeth.f.303. |
| [20] | Jurasinski G, Retzer V, Beierkuhnlein C. Inventory, differentiation, and proportional diversity: a consistent terminology for quantifying species diversity[J]. Oecologia, 2009, 159(1) : 15–26 .DOI:10.1007/s00442-008-1190-z. |
| [21] | D'Argenio V, Casaburi G, Precone V, et al. Comparative metagenomic analysis of human gut microbiome composition using two different bioinformatic pipelines[J]. BioMed Res Int, 2014: 325340.DOI:10.1155/2014/325340. |
| [22] | Khamis A, Raoult D, La Scola B. rpoB gene sequencing for identification of Corynebacterium species[J]. J Clin Microbiol, 2004, 42(9) : 3925–3931 .DOI:10.1128/JCM.42.9.3925-3931.2004. |
| [23] | Claesson MJ, Wang Q, O'Sullivan O, et al. Comparison of two next generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions[J]. Nucleic Acids Res, 2010, 38(22) : e200.DOI:10.1093/nar/gkq873. |