 2017, Vol. 28
2017, Vol. 28扩展功能
文章信息
- 单振菊, 邱德义, 陈健, 岳巧云
- SHAN Zhen-ju, QIU De-yi, CHEN Jian, YUE Qiao-yun
- 二代测序技术检测不同环境下美洲大蠊携带细菌的研究
- The detection of bacteria carried by Periplaneta americana from different environment by next generation sequencing
- 中国媒介生物学及控制杂志, 2017, 28(6): 543-549
- Chin J Vector Biol & Control, 2017, 28(6): 543-549
- 10.11853/j.issn.1003.8280.2017.06.007
-
文章历史
- 收稿日期: 2017-07-10
- 网络出版时间: 2017-10-10 13:59




2 中山大学生命科学学院, 广东 广州 510275
2 College of Life Science Sun Yat-sen University
蜚蠊与人类生活关系密切,是全球性的卫生害虫,可传播多种疾病[1]。因此,对蜚蠊携带的细菌进行研究具有重要意义。蜚蠊携带细菌的检测技术主要有细菌培养、生化鉴定、免疫学技术和探针杂交技术等,各有优缺点。某些自动鉴定系统数据库有限,有时无法准确地鉴定经过长期培养的细菌种类,仍需要做大量的形态学鉴定和生化反应等补充实验[2-4]。非单一菌株16S rRNA的分子鉴定研究需扩增转化后挑单克隆进行,或需先将细菌进行生化分离培养后再鉴定,易造成细菌的漏检,且耗时长、成本高、通量低[5],对实验室的安全级别有一定的要求,而媒介生物携带的细菌不是单一菌株。二代测序技术(NGS)以Illumina公司的Solexa、ABI公司的SOLiD和Roche公司的454技术为代表,具有通量高、准确率高和操作简便等特点[6-8]。近年来,越来越多的研究者开始使用Illumina高通量测序平台检测16S rRNA和16S rDNA的可变区,研究环境微生物的物种丰度、微生物多样性或病畜体内病原体的情况[9-12],但尚无采用NGS研究蜚蠊携带细菌的研究。
细菌的16S rRNA有9个可变区(V1~V9),该可变区在不同种的细菌间序列有一定差异,可用于进行细菌种类的鉴定[13-14]。研究报道,V6区适合鉴定除肠杆菌外其他所有细菌,V4、V5、V7和V8区不能用于种属鉴定[14]。V2、V3高变区可对大多数细菌进行鉴定,但其片段较短[3],V4高变区适合鉴定较复杂的细菌[15]。本研究通过对细菌16S rDNA的9个可变区(V1~V9)进行分析,选出最适合的可变搭配V3~V4可变区作为靶基因,利用NGS尽可能一次性检出1只或几只蜚蠊所携带的未知细菌种类,从而掌握不同环境下蜚蠊携带细菌的基本情况。为快速检测蜚蠊携带细菌提供技术支持,也为地方疾病预防控制部门开展调查提供资料。
1 材料与方法 1.1 样本采集2016年8-9月夜晚在中山地区港口、居民区和医院采集蜚蠊。将样本放入灭菌的离心管中,置-80 ℃冰箱冷冻处死。在生物安全柜内进行形态鉴定后,每一生境选取4~5只美洲大蠊(Periplaneta americana)于-80 ℃保存备用。
1.2 美洲大蠊携带细菌DNA的提取 1.2.1 美洲大蠊体表细菌DNA的提取每一生境各选取4~5只冷冻处死的美洲大蠊,分别置于灭菌离心管中,作为平行。在每个离心管中加入2 ml 0.9% NaCl溶液,漩涡振荡30 s,8 900×g离心3 min,底部留约50 μl,弃去其他上清液至75%乙醇中消毒丢弃,剩余50 μl溶液吹打悬浮后转移到另一只新灭菌的离心管中,重复3次。将收集的菌液95 ℃水浴10 min热裂解细菌,13 400×g离心2 min,收集上清液,4 ℃保存备用。
1.2.2 美洲大蠊体内细菌DNA的提取将1.2.1体表清洗过的美洲大蠊移入装有2 ml 75%乙醇的离心管中浸泡5 min进行消毒处理,重复3次;后加入2 ml 0.9% NaCl,漩涡振荡30 s,清洗表面残余的乙醇,后将样本转移到干净的培养皿中,放置在生物安全柜中晾干。用灭菌的镊子将样本移至新离心管中,将标本腹部研碎成糊状,采用1.2.1的方式收集模板DNA,4 ℃保存备用。
1.3 PCR扩增16S V3~V4可变区的引物:V3-F:5′-CCA GAC TCC TAC GGG AGG CAG-3′;V4-R:5′-GGA CTA CHV GGG TWT CTA AT-3′[16](由Thermo Fisher Scientific Inc广州公司合成)。对提取的美洲大蠊体内、体表细菌DNA进行PCR扩增和琼脂糖凝胶电泳分析。PCR反应体系(100 μl):2×KOD Buffer 50 μl;dNTP(2 mmol/L)5 μl;KOD FX polymerase(Toyobo Life Science Department)2 μl(1 U/μl);引物V3-F(20 mmol/L)2 μl;引物V3-R(20 mmol/L)2 μl;模板DNA 6 μl;加ddH2O补足至100 μl。PCR反应条件:94 ℃预变性2 min。98 ℃变性10 s,45 ℃退火30 s,68 ℃延伸30 s,30个循环。68 ℃延伸10 min,10 ℃保温。
1.4 NGS测序法NGS数据分析参照Bennett等[17]方法。PCR扩增产物送至华大基因有限公司,采用Illumina HiSeqTM2000进行测序,测序策略为PE100。测序流程为回收目的Amplicon片段,用T4 DNA Polymerase、Klenow DNA Polymerase和T4 PNK将打断形成的粘性末端修复成平末端,再通过3′端添加碱基A,使得DNA片段能与3′端带有T碱基的特殊接头连接,下机数据经过数据过滤,滤除低质量的序列,剩余高质量的有效序列方可用于后期分析,通过序列间的重叠关系将序列拼接成目的片段。
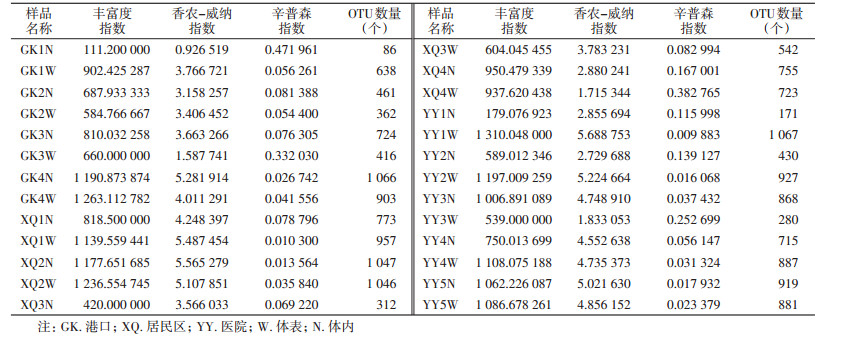
1.5 样品运算分类单位(OTU)及复杂性分析在97%相似度下,将目的片段聚成OTU,统计每个样品中OTU数量。通过mothur(v1.31.2)软件计算样品的Alpha多样性值,并用R(v3.1.1)软件制作相应的稀释曲线图。样品的OTU数值越大,说明该样品的物种越丰富。丰富度指数、香农-威纳指数越大,辛普森指数越小,说明样品中的物种越丰富。
1.6 OTU韦恩分析在97%的相似度下,得到每个样品的OTU,利用韦恩图展示多样品共有和各自特有的OTU数量,直观展示样品间OTU的重叠情况。
1.7 不同环境下美洲大蠊携带细菌种类差异分析 1.7.1 样品Beta多样性热图分析Beta多样性分析是用于比较1对样品的物种多样性差异。分析各种群在样品中的含量,进而计算出不同样品间的Beta多样性值。利用QIIME(v1.80)软件分析Beta多样性。利用R(v3.1.1)软件中NMF包的heatmap制作Beta多样性热图。
1.7.2 样品聚类分析利用QIIME(v1.80)软件进行聚类分析,采用迭代算法,在加权物种分类丰度信息的情况下,使用所有样品中序列数最少的样品序列数的75%进行抽样分析,迭代100次后综合统计得到最终的差异,统计结果得到聚类树,利用R(v3.1.1)软件制图。聚类方法为非加权组平均法(unweighted pair group method with arithmetic mean,UPGMA)。
1.8 样品携带细菌种类及致病性分析通过与数据库进行比对,对OTU进行物种分类,并分别在属、种两个分类等级对各个样品作物种柱状图。根据文献[18]对获得的细菌种类进行致病性分析。
2 结果 2.1 细菌16S rDNA PCR扩增结果利用V3F-V4R引物对美洲大蠊携带的细菌16S rDNA进行扩增,均获得有效的DNA条带,见图 1。

|
| 注:M.分子质量标准;-.阴性对照;1~8.港口样品;9~16.居民区样品;17~26.医院样品 图 1 美洲大蠊体内、体表携带细菌V3F-V4R扩增结果 Figure 1 V3-V4 amplification of bacteria carried by P. americana both inside and outside |
| |
26个样品共产生2 141个OTU,其中YY1W、GK4N、XQ2N和XQ2W样本中物种较丰富,GK1N物种丰富度较小,各样品OTU及生物多样性指数见表 1。由稀释曲线图 2可知,序列数<5 000条,样本的多样性随测序深度增加而迅速增加;序列数为5 000~10 000条时,多样性增加趋于平缓,说明样本的测序量充分,可覆盖所有细菌物种,样品中物种丰度足以满足后续分析。

|
| 注:GK.港口;XQ.居民区;YY.医院;W.体表;N.体内 图 2 美洲大蠊样本丰富度指数稀释曲线 Figure 2 Chao rarefaction curves for samples of P. americana |
| |
由图 3可知,同一个体美洲大蠊体内、体表检出相同细菌种数所占比例最低为C1(医院采集),仅为8.69%,最高为B2(居民区采集),达61.90%。

|
| 注:A1~A4.港口4个平行样品美洲大蠊体内、体表分别检出细菌种类数韦恩分析;A5.港口4个样本体内、体表检出细菌种类数韦恩分析;B1~B4.居民区4个平行样品美洲大蠊体内、体表分别检出细菌种类数韦恩分析;B5.居民区4个样本体内、体表检出细菌种类数韦恩分析;C1~C5.医院5个平行样品美洲大蠊体内、体表分别检出细菌种类数韦恩分析;C6.医院4个样本体内、体表检出细菌种类数韦恩分析。左边圆圈代表体表检出细菌种类数,右边圆圈代表体内检出细菌种类数,重叠部分代表体内、体表检出相同细菌种类数 图 3 港口、居民区、医院美洲大蠊同一个体的体内、体表检出细菌种类数韦恩分析 Figure 3 Venn analysis of bacteria carried by P. americana both inside and outside the body of the same individual collected from port, residential and hospital areas |
| |
由图 4可见,同一地点不同美洲大蠊个体体内、体表检出相同的细菌种类所占比例均较低,居民区美洲大蠊4个不同个体体内、体表携带相同细菌种类分别为124和98种;港口分别为94和16种;医院分别为113和43种。医院(C1、C2)不同美洲大蠊个体间体内、体表携带细菌种类重复较明显,居民区(A1、A2)和港口(B1、B2)不同美洲大蠊个体间体内、体表携带细菌种类重复不明显。

|
| 注:XQ.居民区;GK.港口;YY.医院;W.体表;N.体内 图 4 居民区(A1、A2)、港口(B1、B2)和医院(C1、C2)美洲大蠊不同个体间体内、体表检出细菌种类数韦恩分析 Figure 4 Venn analysis of bacteria carried inside and outside of the body from different individuals P. americana collected from ports, residential, and hospital areas |
| |
利用NGS从港口、居民区和医院采集的美洲大蠊体内、体表检出OTU分别为1 564、1 786和1 779个。3个地点采集的美洲大蠊体内、体表、体内+体表检出相同OTU数分别为891、935和1 242个,见图 5。

|
| 注:XQ.居民区;YY.医院;GK.港口;W.体表;N.体内 图 5 港口、居民区、医院采集的美洲大蠊体内、体表、体内+体表检出细菌种类数韦恩分析 Figure 5 Venn analysis of bacteria carried by P. americana both inside and outside collected from ports, residential, and hospital areas |
| |
对样品进行多样性热图分析和聚类分析发现,β多样性指数越大,样品相似性越低,同一地点样品间和不同地点样品间物种差异不明显(图 6A);同一地点样品未完全聚在一起(图 6B)。两种分析方法均说明美洲大蠊携带的细菌种类与环境无明显区域特异性。

|
| 注:A. Beta多样性热图分析(β多样性指数,按不同地点美洲大蠊体内、体表进行分组);B. UPGMA分析(按不同地点美洲大蠊体内、体表进行分组)。GK.港口;YY.医院;XQ.居民区;W.体表;N.体内 图 6 不同地点美洲大蠊携带细菌种类差异分析 Figure 6 Species variation analysis of bacteria carried by P. americana both inside and outside collected from ports, residential, and hospital areas |
| |
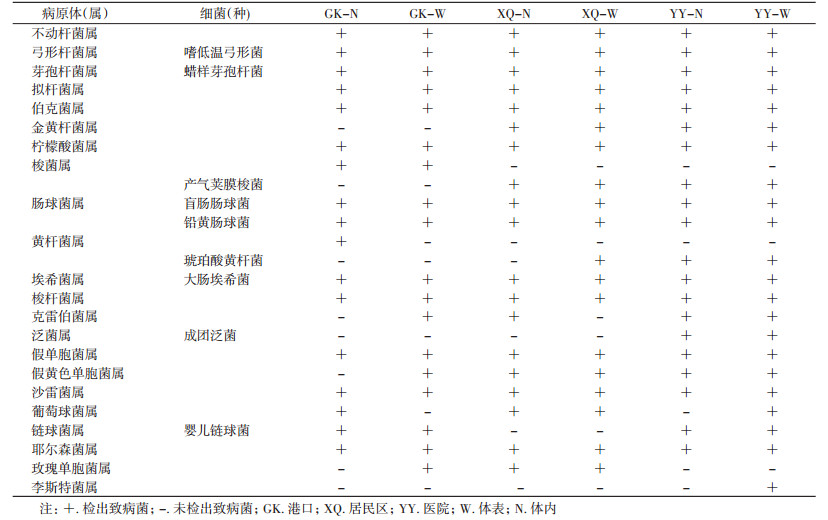
利用NGS从港口、居民区和医院美洲大蠊体内、体表分别可鉴定出细菌123属36种、131属42种和136属48种。从图 7可以直观地看出不同物种在每个样品中所占的比例。将物种丰度在所有样品中均<0.5%的物种全部合并成其他。通过对这些细菌进行致病性分析,港口、医院和居民区检出致病菌分别为18、21和18属,鉴定到种的分别为6、9和8种,见表 2。

|
| 注:GK.港口;XQ.居民区;YY.医院;W.体表;N.体内 图 7 细菌属(A)和种(B)水平上样品物种分布 Figure 7 Species distribution bar graph of samples at genus (A) and species (B) level |
| |
 |
本研究通过对16S rDNA V1~V9可变区的分析,选取V3~V4可变区作为目的基因,建立了一次性检测蜚蠊携带细菌的方法。该方法省去细菌培养、挑单克隆等操作,避免了因非单一菌株16S rRNA的分子鉴定扩增转化后挑单克隆时漏检菌落及操作过程易污染等问题。
与传统方法相比较,NGS方法操作简便,效率高,可实现细菌一次性的高通量检测。部分学者利用传统方法从美洲大蠊体内和体表检出25个属的细菌[1],而NGS法可检出100多个属的细菌,大大提高了检出种类。且利用NGS方法检测蜚蠊携带的细菌可获得大量的数据信息,便于对不同样品间的关系进行生物学分析研究,如样品间的OTU主成分分析、多样性热图分析和聚类分析等,而传统方法则无法实现。
通过对同一环境和不同环境采集的美洲大蠊携带细菌的种类进行分析,结果表明,不同环境下美洲大蠊携带的细菌种类与环境的关系不明显。在今后研究中应加强每个地点的采样数量,并以采样环境存在的细菌为参照进一步分析。本研究的3个采样地点均检出大量的细菌,其中包括部分致病菌和条件致病菌。目前,由于大量使用抗生素,较多细菌产生了耐药性,如不动杆菌属、克雷伯菌属、大肠埃希菌、沙雷菌属和变形杆菌属等,已被WHO列为“抗生素耐药重点病原体”清单,这些细菌种族严重威胁人类健康[19]。尤其在医院环境下,蜚蠊作为医学细菌等重要微生物的传播载体应引起足够重视。
随着我国对外贸易活动增加,医学媒介生物及其虫媒传染病的潜在风险性与日俱增。因此,应积极开展医学媒介生物及其携带细菌的检测和控制,从而有效地防止传染病的传播,保护人类健康。同时可作为有力的卫生检疫技术壁垒为我国对外经济贸易服务,是全面提升我国卫生检疫工作水平的重要突破口和创新点,应给予高度重视。
| [1] |
徐毅. 蜚蠊病原体携带研究进展[J]. 实用预防医学, 2015, 22(5): 636-639. |
| [2] |
邓梅葵, 孙迎, 韩雯晴. 细菌鉴定方法[J]. 生物医学工程学进展, 2014, 35(2): 84-88. |
| [3] |
Becker K, Harmsen D, Mellmann A, et al. Development and evaluation of a quality-controlled ribosomal sequence database for 16S ribosomal DNA-based identification of Staphylococcus species[J]. J Clin Microbiol, 2004, 42(11): 4988-4995. DOI:10.1128/JCM.42.11.4988-4995.2004 |
| [4] |
Bosshard PP, Zbinden R, Abels S, et al. 16S rRNA gene sequencing versus the API 20 NE system and the VITEK 2 ID-GNB card for identification of nonfermenting Gram-negative bacteria in the clinical laboratory[J]. J Clin Microbiol, 2006, 44(4): 1359-1366. DOI:10.1128/JCM.44.4.1359-1366.2006 |
| [5] |
Capurro A, Artursson K, Waller KP, et al. Comparison of a commercialized phenotyping system, antimicrobial susceptibility testing, and tuf gene sequence-based genotyping for species-level identification of coagulase-negative Staphylococci isolated from cases of bovine mastitis[J]. Vet Microbiol, 2009, 134(3/4): 327-333. |
| [6] |
Quail MA, Kozarewa I, Smith F, et al. A large genome center's improvements to the Illumina sequencing system[J]. Nat Med, 2008, 5(12): 1005-1010. |
| [7] |
Meyer M, Stenzel U, Hofreiter M. Parallel tagged sequencing on the 454 platform[J]. Nat Protoc, 2008, 3(2): 267-278. DOI:10.1038/nprot.2007.520 |
| [8] |
Mardis ER. The impact of next-generation sequencing technology on genetics[J]. Trends Genet, 2008, 24(3): 133-141. DOI:10.1016/j.tig.2007.12.007 |
| [9] |
Degnan PH, Ochman H. Illumina-based analysis of microbial community diversity[J]. ISME J, 2012, 6(1): 183-194. DOI:10.1038/ismej.2011.74 |
| [10] |
Gilbert JA, Meyer F, Antonopoulos D, et al. Meeting report:the terabase metagenomics workshop and the vision of an earth microbiome project[J]. Stand Genomic Sci, 2010, 3(3): 243-248. DOI:10.4056/sigs.1433550 |
| [11] |
Bartram AK, Lynch MDJ, Stearns JC, et al. Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads[J]. Appl Environ Microbiol, 2011, 77(11): 3846-3852. DOI:10.1128/AEM.02772-10 |
| [12] |
Xiao M, Zhang ZZ, Wang JX, et al. Bacterial community diversity in a low-permeability oil reservoir and its potential for enhancing oil recovery[J]. Bioresour Technol, 2013, 147: 110-116. DOI:10.1016/j.biortech.2013.08.031 |
| [13] |
Chakravorty S, Helb D, Burday M, et al. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria[J]. J Microbiol Methods, 2007, 69(2): 330-339. DOI:10.1016/j.mimet.2007.02.005 |
| [14] |
Moreau MM, Eades SC, Reinemeyer CR, et al. Illumina sequencing of the V4 hypervariable region 16S rRNA gene reveals extensive changes in bacterial communities in the cecum following carbohydrate oral infusion and development of early-stage acute laminitis in the horse[J]. Vet Microbiol, 2014, 168(2/4): 436-441. |
| [15] |
Claesson MJ, Wang Q, O'Sullivan O, et al. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions[J]. Nucleic Acids Res, 2010, 38(22): e200. DOI:10.1093/nar/gkq873 |
| [16] |
Jin CY, Zeng ZY, Fu ZW, et al. Oral imazalil exposure induces gut microbiota dysbiosis and colonic inflammation in mice[J]. Chemosphere, 2016, 160: 349-358. DOI:10.1016/j.chemosphere.2016.06.105 |
| [17] |
Bennett DC, Tun HM, Ji EK, et al. Characterization of cecal microbiota of the emu (Dromaius novaehollandiae)[J]. Vet Microbiol, 2013, 166(1/2): 304-310. |
| [18] |
杨正时, 房海. 人及动物病原细菌学[M]. 石家庄: 河北科学技术出版社, 2003, 2-180.
|
| [19] |
WHO. 世卫组织发布迫切需要新型抗生素的细菌清单[EB/OL]. (2017-02-27)[2017-04-05]. http://www.who.int/mediacentre/news/releases/2017/bacteria-antibiotics-needed/zh/.
|