 2016, Vol. 27
2016, Vol. 27扩展功能
文章信息
- 韩娜, 张琳, 张雯, 强裕俊, 侯学霞, 陈晨, 郝琴, 张媛媛
- HAN Na, ZHANG Lin, ZHANG Wen, QIANG Yu-jun, HOU Xue-xia, CHEN Chen, HAO Qin, ZHANG Yuan-yuan
- 长角血蜱细菌群落结构及多样性研究
- Studies on the composition and diversity of the bacterial community in Haemaphysalis longicornis
- 中国媒介生物学及控制杂志, 2016, 27(5): 426-431
- Chin J Vector Biol & Control, 2016, 27(5): 426-431
- 10.11853/j.issn.1003.8280.2016.05.002
-
文章历史
- 收稿日期: 2016-05-23
- 网络出版时间: 2016-08-11




2 感染性疾病诊治协同创新中心, 浙江 杭州 310003;
3 首都医科大学附属北京地坛医院传染病研究所, 新发突发传染病研究北京市重点实验室, 北京 100015
2 Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases;
3 Institute of Infectious Diseases, Beijing Ditan Hospital, Capital Medical University, Beijing Key Laboratory of Emerging Infectious Diseases
蜱是一类可在脊椎动物(包括人类)体表寄生并吸血的节肢动物,吸血时释放毒素,严重威胁人类健康并给畜牧业造成严重的经济损失;同时,其从宿主获取病原体,在体内发育繁殖,且可经卵、变态期传递,可长期储存病原微生物。蜱在我国分布较广泛,且种群数量大,是仅次于蚊类的第二大人畜共患病的传播媒介和宿主[1]。近30年,随着诊断识别技术的发展,目前已报道蜱传疾病病原体多达31种,其中14种可感染人类,包括多种病毒、细菌、螺旋体、立克次体、血液原虫及衣原体等,而未知、新发的蜱传疾病病原体仍不断被检出[2]。因此,对蜱自然携带细菌的群落结构及种群多样性进行分析可对自然疫源地主要存在的细菌进行本底调查,为快速、准确地检出、发现病原体,以及对临床诊断、检测及控制暴发流行有重要的指导意义。
由于技术方法的限制,以往蜱携带菌群的研究不够深入。经典的细菌分离培养方法繁琐、耗时长、成本高,对操作人员的经验和技术要求较高;且一些细菌在实验室条件下较难培养,大大降低了细菌的检出率;目前,核酸检测方法更为普遍,常规的PCR与荧光定量PCR、核酸杂交及在此基础上发展起来的基因芯片等方法,均依赖于已知细菌的基因组序列,对于检测新发甚至完全未知的病原体存在很大的局限性。宏基因组学的兴起和近年来发展迅速的二代测序技术可同时对上百份样本进行高通量核酸分子测序,不依赖于细菌的分离培养,尤其适合从混杂的环境样本或临床样本中发现重要的病原微生物,成为鉴定未知病原菌较为有效的技术手段[3]。本研究通过对辽宁省24份野外采集的长角血蜱(Haemaphysalis longicornis)样本携带细菌的16S rDNA基因V3~V4区段进行高通量测序,对蜱自然携带的虫媒微生物进行筛查,研究蜱携带的微生物群落结构,快速、准确地了解自然疫源地主要存在的虫媒微生物种类,评价物种丰富度及均匀度,为进一步探讨蜱与疫源地、病原体间关系提供依据。
1 材料与方法 1.1 样本采集2013年7月在辽宁省抚顺市岗山地区利用布旗法采集蜱,按蜱种分类鉴定为长角血蜱,10~15只为1管置于液氮内保存。
1.2 仪器和试剂QIAamp DNA Stool Mini Kit基因组提取试剂盒(德国QIAGEN公司);NanodropTM 2000/2000C超微量分光光度计(美国Thermo Scientific 公司);PCR引物由生工生物工程(上海)股份有限公司合成;Miseq测序仪(美国Illumina公司)。Q5® Hot Start High-Fidelity 2×Master Mix PCR扩增试剂(英国NEB公司)。文库构建:Kapa HTP Library Prep Kit Illumina(美国KAPA公司);文库定量:Quant-iTTM PicoGreen dsDNA Assay Kit(美国ThermoFisher公司)。
1.3 细菌DNA提取对野外采集的24组蜱样本体表进行消毒,用75%乙醇浸泡3次,每次10 min。随后用PBS漂洗3次,每次5 min,风干。将消毒后的蜱置于1.5 ml Eppendoff离心管中,加入液氮反复研磨3~5次。基因组DNA提取按QIAamp DNA Stool Mini Kit试剂盒说明操作,-20 ℃保存备用。
1.4 细菌16S rDNA基因V3~V4区高通量测序 1.4.1 PCR引物及反应条件以提取的基因组DNA为模板,对样本中总细菌基因组16S rDNA基因的V3~V4高变区进行扩增,通用引物上游:Bakt_341F (5′-CCT ACG GGN GGC WGC AG-3′);下游:Bakt_805R (5′-GAC TAC HVG GGT ATC TAA TCC-3′),目的片段长度约为460 bp。PCR反应体系共50 μl,包括2×Taq PCR Mix聚合酶 25 μl,DNA模板4 μl,0.5 μmol/L上下游引物各0.2 μl,ddH2O补足至50 μl。PCR反应条件:95 ℃预变性5 min;然后进行30个循环,每个循环包括:95 ℃变性40 s,58 ℃退火40 s,72 ℃延伸60 s;最后一个循环结束后72 ℃延伸7 min。PCR产物经NanoDrop检测浓度,1%琼脂糖凝胶电泳及凝胶成像系统检测其目的片段。
1.4.2 文库构建和高通量测序将上述PCR产物进行纯化后,按照文库构建试剂盒操作步骤构建小片段文库,使用核酸定量试剂盒测定文库浓度,并利用Illumina Miseq高通量测序平台进行双末端测序,测序策略为PE300,reads数≥30 000条。
1.4.3 生物信息学分析使用自主编写perl脚本对下机数据进行分样、质量控制。用PEAR软件将双端序列拼接、过滤成Tags。使用QIIME V1.9.1软件将Tags进行分类单元(operational taxonomic units,OTUs)聚类、分析和物种注释,通过计算多样性指数和绘制稀疏曲线进行样本复杂度分析(α-多样性);细菌群落结构和物种多样性,通过主坐标分析(principal coordinate analysis,PCoA)及UPGMA聚类分析进行细菌群落结构的比较分析(β-多样性),寻找组别相关菌群或菌种。
1.5 统计学分析计量资料采用(x±s)表示。利用R软件及GraphPad Prism 5.0软件进行统计学分析及绘图。
2 结果 2.1 长角血蜱的16S rDNA基因测序结果为提高16S rDNA 鉴定微生物种属的灵敏性和特异性,使用通用引物和Illumina Miseq PE300双端测序扩增及测序16S rDNA 的V3~V4区段。24组蜱样本共获得高质量拼接Tags 1 070 624条,但其中tick24、tick14和tick22样本获得Tags数<5 000(分别为4 413、388和22),序列数过少无法客观地反映蜱在自然状态下携带的微生物群落结构,后续分析中将去除该3个样本。其余21个样本中平均测序reads为50 752条,序列的长度为444.96 bp,(G+C)含量为53.44%。各样本测序结果见表 1。
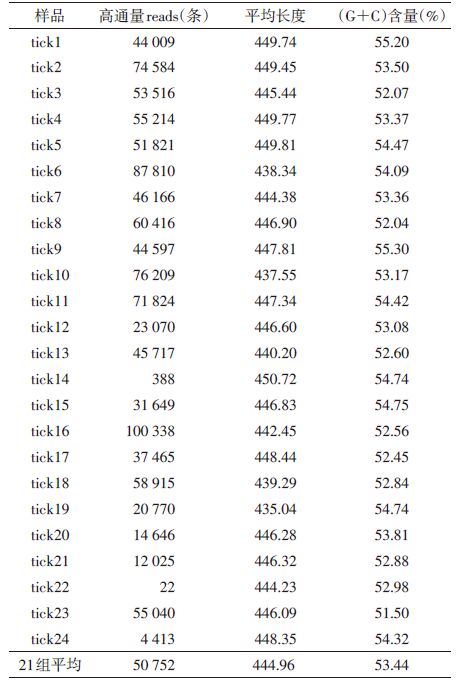
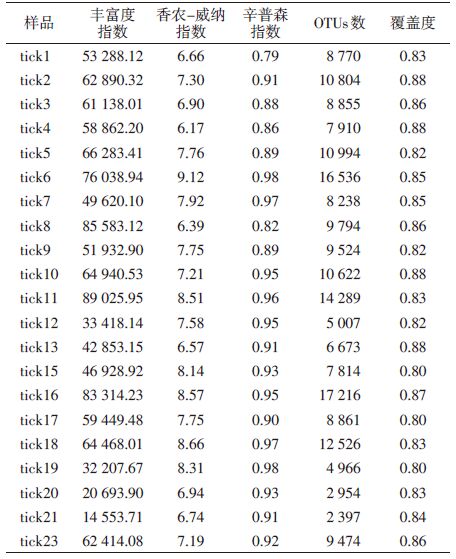
21组长角血蜱样本共获得 145 479个OTUs,每组样本获得(9 248±3 843)个OTUs。样本的丰富度指数为56 185.95±19 830.94,覆盖度为0.84±0.03,多样性指数(Simpson/Shannon-Weiner)分别为(0.92±0.05)和(7.53±0.82),见表 2。从稀疏曲线看,在<5 000条reads时,样本的多样性随测序深度增加而迅速增加;在5 000~15 000条序列时,多样性增加缓慢,之后为平台期,见图 1。各样本稀疏曲线证明测序量充分,可覆盖所有细菌物种,说明样本中的物种丰富度足以满足后续分析。

|
| 图 1 21组长角血蜱样本携带微生物菌群的稀疏曲线 Figure 1 Shannon rarefaction curves for 21 H. longicornis ticks |
| |
在门水平,21组样本携带的相关细菌群落隶属于16个门,前4位为变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)、放线菌门(Actinobacteria)和拟杆菌门(Bacteroidetes),占总菌群的94.06% 以上。变形菌门所占比例最大,变化范围及相对丰度变异范围也比较广,为46.07%~97.27%,成为蜱优势菌群(图 2a)。在门水平检测到古细菌广古菌门,但相对丰度<0.01%。各样本在门水平具有各自的分布特征,每组样本均有0.5%~5.0%的reads无明确分类。

|
| 注: a. 门水平群落结构; b. 纲水平,变形菌门下各纲群落结构; c. 属水平群落结构; 图中相对丰度<0.01%物种的颜色不明显。 图 2 21组长角血蜱样本微生物群落结构 Figure 2 The bacterial community structure of 21 H. longicornis ticks |
| |
在纲水平,相对丰度颇高的变形菌门进一步划分为4个纲,根据相对丰度依次为γ-变形菌纲(Gammaproteobacteria,14.63%~86.09%)、α-变形菌纲(Alphaproteobacteria,3.11%~45.63%)、β-变形菌纲(Betaproteobacteria,1.57%~23.91%)和δ-变形菌纲(Deltaproteobacteria,0~0.06%)(图 2b)。
在属水平,微生物群落隶属于383属。相对丰度位于前20位的属所占比例>74.44%(图 2c)。蜱样本寄生的微生物在属水平有各自的分布特征,个体间变异也较大。Enterobacteriaceae_Other代表肠杆菌科未明确分属的一类细菌,在多组样本中所占比例较大,在tick1中高达79.32%。柯克斯体属(Coxiella)在21组样本中均存在,在tick3、tick8、tick12和tick7样本比例高达29.45%~60.81%;寡养单胞菌属(Stenotrophomonas)在tick4、tick20中相对丰度高达44.82%;不动杆菌属(Acinetobacter)在tick6、tick10、tick16和tick18中比例为11.42%~19.72%;假单胞菌属(Pseudomonas)在tick4、tick5和tick21中比例为13.28%~44.58%;立克次体属(Rickettsia)在11组蜱样本中存在,但在tick13中相对丰度最高,达29.33%,在其余10组样本中比例均<0.01%。
2.4 蜱群落差异分析非加权组平均法(UPGMA)分析结果显示21组蜱样本可聚成2个大簇,即GⅠ和GⅡ簇,每簇中蜱样本≥3个(图 3a)。基于UniFrac距离绘制的主坐标图(PCoA)同样展示了2个簇的存在(图 3b)。tick2、tick4、tick16和tick23则比较分散,与大簇间的距离较远,且每个仅1组样本,故暂未将其划分为独立簇。比较GⅠ和GⅡ在属水平的菌群结构,发现共有属群40个(图 3c)。GⅠ特有属群21个,包含Kocuria、Chryseobacterium、Wautersiella、Macrococcus、Mycoplana、Enterobacter、Erwinia、Klebsiella、Salmonella、Enhydrobacter等属;GⅡ特有属群11个,包含Brevibacterium、Brachybacterium、Exiguobacterium、Rummeliibacillus、Rhodoplanes、Nitratireductor等属。

|
| 注: a. UPGMA分析; b. 基于UniFrac距离的PCoA分析; c. GⅠ和GⅡ属水平Venn分析。 图 3 长角血蜱携带微生物的菌群群落差异分析 Figure 3 The microbiome variation analysis of H. longicornis ticks |
| |
本研究采用16S rDNA高通量测序获得野外长角血蜱自然携带的微生物优势菌群依次为变形菌门、厚壁菌门、放线菌门和拟杆菌门,与鹿蜱(Ixodes scapularis)[4]、血红扇头蜱(Rhipicephalus sanguineus)、图兰扇头蜱(Rh. turanicus)[5]及其他昆虫中的研究结果一致[6-8]。其中变形菌门相对丰度高达46.07%~97.27%,可能与变形菌门细菌在自然界中分布广泛有关。作为细菌中最大的一门,变形菌门包括诸多病原菌,如大肠埃希菌(Escherichia coli)、沙门菌(Salmonella)、弧菌(Vibrio)和螺杆菌(Helicobacterpylori)等[9]。变形菌门中γ-变形菌纲的相对丰度最高,提示其易在蜱体内生存。拟杆菌门参与碳水化合物发酵、多糖代谢、胆汁酸和类固醇等养分代谢,维持正常的生理功能和微生态平衡,对长角血蜱有重要意义[9-10]。
群落结构及多样性指数分析得出长角血蜱个体间携带的微生物种类和相对丰度差异较大,与个体的摄食习惯、雌雄体及不同的生长阶段有关,可能是采自同一地区的蜱携带菌群差异的原因。唐昊等[11]发现不同阶段的雌雄微小牛蜱(Boophilus microplus)中肠内容物的细菌种群结构存在一定程度的差别。 但若将在所有样本或至少在大多数样本中检测到的丰度较高的微生物属定义为蜱的“核心微生物群”,则本研究中检测到的前20个属,除立克次体属外,其余19个属在21组样本中均存在,且相对丰度较高,可定义为长角血蜱的“核心微生物群”。
通过UPGMA和PCoA分析长角血蜱的微生物群落虽呈多样性,但可形成不同的“蜱簇”。除共有菌群外,GⅠ和GⅡ蜱簇均具有特有菌群。tick2、tick4、tick16和tick23游离在蜱簇外,可能与研究取样量偏小有关。目前已发现蜱800余种,其自然携带的微生物应是巨大的微生物资源库,随着取样量的扩大,可能会呈现更多的蜱簇。
通过对蜱携带微生物菌群的结构分析,发现菌群中存在大量致病菌和条件致病菌,如肠杆菌属(Enterobacter spp.)、柯克斯体属、寡养单胞菌属、立克次体、无形体(Anapasma)、不动杆菌属、假单胞菌属、克雷伯菌属(Klebsiella)、变形杆菌属(Proteus)、沙门菌中的一些种类,在一定条件下可以引起人类和动物致病。柯克斯体属广泛存在于长角血蜱样本中,在tick17样本中相对丰度高达47.91%,其下的Coxiella burnetii是引起Q立克次体热的病原体。寡养单胞菌属为条件致病菌,主要引起院内感染,在非发酵菌引起的感染中,仅次于铜绿假单胞菌(Pseudomonas aeruginosa)和鲍曼不动杆菌(Acinetobacter baumannii),而居临床分离阳性率的第3位。52.38%(11/21)的样本检出立克次体,大多数样本相对丰度<0.01%,但tick13高达29.33%,具体原因未知。在5组样本中检出无形体,该菌是人粒细胞无形体病病原体。仅1组样本中检出衣原体。除常见的致病菌外,短波单胞菌属(Brevunmdimonas)和代尔夫特菌属(Delftia)均以较高丰度广泛存在于蜱中,一般不致病,但也有报道其存在于免疫力低下的患者中,如癌症患者[12]。本研究中未检测到螺旋体和支原体(Mycoplasma)。基于16S rDNA高通量测序证实了古细菌的存在,但相对丰度较低,主要为甲烷袋状菌属(Methanoculleus)和嗜盐菌科(Halobacteriaceae)的古细菌,可能与蜱特殊的生存环境相关。
我国地貌形态复杂多样,境内河湖交错,生态景观多样,气候类型复杂,适合蜱孳生繁衍。长角血蜱与疾病密切相关,且种群密度较高,为辽宁省的优势蜱种之一。随着蜱叮咬引起不明原因的未知、新发传染性疾病的出现及旅游、野外活动的增加,蜱传疾病逐渐被重视,对其携带病原体的检测能力也不断增强。通过高通量测序技术对野外蜱寄生菌群进行调查,有助于阐明蜱与其共生微生物间的相互关系,为防控蜱及其相关疾病奠定基础。
| [1] | Jongejan F, Uilenberg G. The global importance of ticks[J]. Parasitology, 2004, 129 (Suppl:S) : 3–14 . |
| [2] | Yu ZJ, Wang H, Wang TH, et al. Tick-borne pathogens and the vector potential of ticks in China[J]. Parasit Vectors, 2015, 8 (1) : 24.DOI:10.1186/s13071-014-0628-x. |
| [3] | Manichanh C, Chapple CE, Frangeul L, et al. A comparison of random sequence reads versus 16S rDNA sequences for estimating the biodiversity of a metagenomic library[J]. Nucl Acids Res, 2008, 36 (16) : 5180–5188 .DOI:10.1093/nar/gkn496. |
| [4] | Benson MJ, Gawronski JD, Eveleigh DE, et al. Intracellular symbionts and other bacteria associated with deer ticks (Ixodes scapularis) from Nantucket and Wellfleet, Cape Cod, Massachusetts[J]. Appl Environ Microbiol, 2004, 70 (1) : 616–620 .DOI:10.1128/AEM.70.1.616-620.2004. |
| [5] | Lalzar I, Harrus S, Mumcuoglu KY, et al. Composition and seasonal variation of Rhipicephalus turanicus and Rhipicephalus sanguineus bacterial communities[J]. Appl Environ Microbiol, 2012, 78 (12) : 4110–4116 .DOI:10.1128/AEM.00323-12. |
| [6] | Corby-Harris V, Pontaroli AC, Shimkets LJ, et al. Geographical distribution and diversity of bacteria associated with natural populations of Drosophila melanogaster[J]. Appl Environ Microbiol, 2007, 73 (11) : 3470–3479 .DOI:10.1128/AEM.02120-06. |
| [7] | Gupta AK, Nayduch D, Verma P, et al. Phylogenetic characterization of bacteria in the gut of house flies (Musca domestica L.)[J]. FEMS Microbiol Ecol, 20152, 79 (3) : 581–593 . |
| [8] | Campbell CL, Mummey DL, Schmidtmann ET, et al. Culture-independent analysis of midgut microbiota in the arbovirus vector Culicoides sonorensis (Diptera: Ceratopogonidae)[J]. J Med Entomol, 2004, 41 (3) : 340–348 .DOI:10.1603/0022-2585-41.3.340. |
| [9] | 布坎南. 伯杰细菌鉴定手册[M]. 8版. 中国科学院微生物研究所,译. 北京:科学出版社,1984:729-735. |
| [10] | Sears CL. A dynamic partnership: celebrating our gut flora[J]. Anaerobe, 2005, 11 (5) : 247–251 .DOI:10.1016/j.anaerobe.2005.05.001. |
| [11] | 唐昊, 赵瑜, 廖芷卉, 等. 微小牛蜱中肠内容物菌群结构分析[J]. 中国病原生物学杂志,2015,10 (6) :487–490. |
| [12] | Han XY, Andrade RA. Brevundimonas diminuta infections and its resistance to fluoroquinolones[J]. J Antimicrob Chemother, 2005, 55 (6) : 853–859 .DOI:10.1093/jac/dki139. |