 2015, Vol. 26
2015, Vol. 26扩展功能
文章信息
- 王海峰, 杨顺林, 周松, 史献明, 胡乐乐, 刘合智, 梁莹
- WANG Hai-feng, YANG Shun-lin, ZHOU Song, SHI Xian-ming, HU Le-le, LIU He-zhi, LIANG Ying
- 河北省鼠疫耶尔森菌多位点串联重复序列分析
- MLVA analysis on Yersinia pestis isolated from plague foci in Hebei province, China
- 中国媒介生物学及控制杂志, 2015, 26(2): 141-144
- Chin J Vector Biol & Control, 2015, 26(2): 141-144
- 10.11853/j.issn.1003.4692.2015.02.008
-
文章历史
- 收稿日期:2014-10-13




2 中国疾病预防控制中心传染病预防控制所
2 National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention
河北省北部与内蒙古相接,南面与河南、山东省毗连,西面与山西省相邻,东临渤海,东北与辽宁省相邻,环抱北京与天津市,交通发达,具有重要的地理位置[1]。1971年12月,河北省首次从长爪沙鼠(Meiionesunguiculataus)体内分离到鼠疫菌,证实了鼠疫疫源地的存在。期间共发生4次动物鼠疫流行,菌型均为鄂尔多斯高原型[2]。鉴于河北省地理位置的特殊性和鼠疫的严重危害性,深入研究分析该疫源地鼠疫菌的分子遗传特征,具有十分重要的意义。多位点串联重复序列分析(MLVA)首先被国外学者引入鼠疫领域[3],利用鼠疫菌基因组中存在不同数目的串联重复序列(VNTR)位点,将鼠疫菌进行分型。张晓嫒[4]和付秀萍等[5]曾利用所公布的VNTR位点将鼠疫菌进行分型。由于我国鼠疫菌类型复杂,需要选用更适宜的VNTR位点进行分析。通过对鼠疫菌全基因组序列的比对,本研究采用15个VNTR位点对河北省长爪沙鼠鼠疫疫源地的116株鼠疫菌进行了MLVA,建立该疫源地鼠疫菌的遗传学档案资料,为鼠疫防控提供依据。
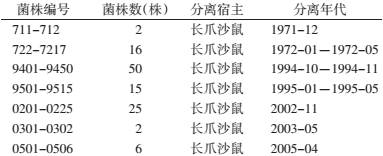
1 材料与方法 1.1 菌株来源实验所用116株鼠疫菌均分离于河北省鼠疫疫源地。1971-1972年分离18株,1994-1995年分离65株,2002-2003年分离27株,2005年分离6株(表 1)。
PCR扩增仪(杭州博日科技有限公司)、高速台式离心机(Eppendorf 5424)、水平电泳仪(北京市六一仪器厂)、凝胶成像仪(上海基因有限公司)。
1.3 试剂DNA提取试剂盒购自Qiagen公司,PCR扩增试剂购自北京全式金生物技术有限公司,DL500 Marker购自大连宝生物工程有限公司,100 bp Marker购自北京赛百盛基因技术有限公司,琼脂糖购自西班牙Biowest。
1.4 方法 1.4.1 引物设计引物MLV1~MLV9、MLV11、MLV12、MLV14为中国疾病预防控制中心(CDC)传染病预防控制所设计提供。引物M52、M59和M61从已发表文献中筛选。本研究使用的引物序列信息见表 2。
冻干菌株经营养琼脂培养基28 ℃ 24~48 h复苏,分离培养,做噬菌体实验,Qiagen试剂盒提取DNA。
1.4.3 PCR反应体系和条件去离子水将提取的鼠疫菌DNA模板10倍稀释,采用25 μl反应体系,其中Supermix 12.5 μl,上、下游引物各1 μl,模板1 μl,去离子水9.5 μl。引物MLV1~MLV4的PCR反应条件:94 ℃预变性5 min,94 ℃变性45 s,60 ℃退火45 s,72 ℃延伸2 min。35个循环。72 ℃延伸5 min。其余11对引物延伸时间为1 min,其他条件同上。
1.4.4 电泳条件引物MLV1~MLV4的扩增产物采用3%琼脂糖凝胶,其余为2%。120 V,40 min。
1.4.5 扩增片段长度分析利用凝胶成像仪分析系统分析,挑选个别反应进行测序,最后确定扩增长度和重复数目。
2 结 果 2.1 同一引物基因扩增菌株结果同一引物所扩增的不同菌株目标条带长度在同一位置,随机挑选出2个反应由生工生物工程(上海)股份有限公司测序,证实同一引物扩增的产物其长度相同,经MegAlign软件和NCBI在线Blast与CO92基因序列比对得出最后的VNTR重复数(表 3)。
 |
分别用中国CDC传染病预防控制所提供的15对引物,针对随机选取不同年代分离的菌株进行扩增,所有引物均扩增出目标条带(图 1~3),且针对不同的VNTR位点,产生各自的重复数目。

|
| 图 1 引物 MLV4对不同菌株的扩增结果(长度为 732 bp) Figure 1 The amplification result of different strains with primer MLV4 |

|
| 图 2 引物 MLV1~MLV4分别对 728、9508、0204、0301、0501 菌株扩增结果 Figure 2 The implication results of the strain 728,9508,0204,0301,0501 with MLV1,MLV2,MLV3,and MLV4 |

|
| 图 3 引物 MLV5~M61 分别对 712、9406两株菌株的扩增结果 Figure 3 The amplification result of the strain 712,9406with primer MLV5-M61 |
每对引物均挑选2个扩增结果进行测序,然后采用Blast软件与CO92比对,确定扩增反应的核心序列重复数目,如图 4所示,VNTR位点M59在CO92的重复数目比测序菌株的重复数目少1个,其余各位点核心序列重复数目见表 3。

|
| 图 4 M59引物扩增产物序列与 CO92比较 Figure 4 Contrast of the sequence in amplification with M59 to CO92 |
实验显示,同一引物针对116株鼠疫菌所得的重复序列均相同,可见分离自河北省境内的菌株同属于一个MLVA型。
3 讨 论MLVA技术在国内被广泛应用于鼠疫菌的基因分型,Zhang等[6]引用并筛选的10个VNTR位点将我国各个疫源地的鼠疫菌株分为74个基因型,并证明基因型与生态型相吻合;Li等[7]利用14+12个VNTR,将我国鼠疫菌进行了更详尽的遗传进化分析;朱俊洁等[8]筛选出5个VNTR位点将云南省鼠疫疫源地的鼠疫菌分为5个基因型。研究显示MLVA是一种可靠的分型方法,不仅可以将我国鼠疫菌进行科学客观的分型,同时也为研究彼此的亲缘关系和遗传进化特征提供帮助。
河北省长爪沙鼠鼠疫疫源地鼠疫菌生态分型为鄂尔多斯高原型[9]。在本研究中,针对同一VNTR位点所设计的引物,所有菌株的重复数目均相同,即同属于一个基因型,与其生态型分型相吻合。尽管该疫源地自1972年以来发生过4次较大的动物鼠疫流行,但其鼠疫菌MLVA遗传特征仍然是稳定的。本研究通过所获得的数据,建立了河北省鼠疫菌MLVA数据库,这些将对未来的鼠疫疫情调查和应急处理提供技术支持。
河北省鼠疫菌株在生物分型中属于中世纪型[10],也即甘油阳性而脱氮实验阴性,在本研究中,同一菌株不同引物扩增的长度和重复数目,有些与CO92不同,也即生物型不同其基因型也不同。本研究通过与全国鼠疫菌MLVA比对,可进一步对河北省鼠疫疫源地菌株进化过程分析提供依据。
将来自于内蒙古长爪沙鼠鼠疫疫源地的菌株99082、99103与河北省菌株比较,发现就同一引物扩增,目标条带并不在相同的位置,显示尽管它们为同一疫源地的鄂尔多斯型菌株,但是仍然在进化过程中有一些不同的变化,这与之前的生化实验结果不一致,分离自两地的菌株其对蜜二糖和麦芽糖的酵解能力也不同[9],可见MLVA更能详尽准确地将菌株分型并做进化分析。
| [1] | 李振海, 陈永明, 李庆芬. 河北省蚤类区划的研究[J]. 医学动物防制, 2006, 22(9):627-632. |
| [2] | 刘合智, 刘满福, 李玉贵. 河北省鼠疫自然疫源地内自然染疫蚤的研究[J]. 中国媒介生物学及控制杂志, 2005, 16(3):206-208. |
| [3] | Klevytska AM,Price LB,SchuppJM,etal. Identificationand characterization of variable-number tandem repeats in the Yersinia pestis genome[J]. J Clin Microbiol, 2001, 39(9): 3179-3185. |
| [4] | 张晓嫒. 中国鼠疫耶尔森菌多位点可变数目串联重复序列分析[D]. 北京:中国疾病预防控制中心, 2008. |
| [5] | 付秀萍, 俞东征, 海荣. 利用M28串联重复序列对鼠疫菌进行基因分型[J]. 疾病控制杂志, 2006, 10(1):21-23. |
| [6] | Zhang XA, Hai R, WeiJC, et al.MLVAdistributioncharacteristics of Yersinia pestis in China and the correlation analysis[J]. BMC Microbiol, 2009, 9:205. |
| [7] | Li YJ,Cui YJ,Cui BZ,etal.Featuresofvariablenumberof tandem repeats in Yersinia pestis and the development of a hierarchical genotyping scheme[J]. PLoS One, 2013, 8(6):e66567. |
| [8] | 朱俊洁, 王鹏, 张蓉, 等. 云南省鼠疫耶尔森菌多位点可变数目串联重复序列分析[J]. 疾病监测, 2013, 28(10):848-852. |
| [9] | 董国润, 王桂琴, 白晓英, 等. 115株鼠疫菌的生物学特性及流行病学研究[J]. 中国媒介生物学及控制杂志, 2001, 12(2):112-114. |
| [10] | 纪树立, 张海峻, 刘云鹏, 等. 我国鼠疫菌分型及其生态学、流行病学意义[J]. 中国地方病学杂志, 1982, 6(5):257-262. |