 2024, Vol. 35
2024, Vol. 35扩展功能
文章信息
- 管毓威, 罗小龙, 周敬祝, 郑德阳, 邓小生, 胡勇, 梁文琴
- GUAN Yu-wei, LUO Xiao-long, ZHOU Jing-zhu, ZHENG De-yang, DENG Xiao-sheng, HU Yong, LIANG Wen-qin
- 贵州省部分地区微小扇头蜱微生物群落多样性及抗生素抗性基因的宏基因组分析
- Metagenomic analysis of microbial community diversity and antibiotic resistance genes of Rhipicephalus microplus in some areas of Guizhou Province, China
- 中国媒介生物学及控制杂志, 2024, 35(4): 394-400, 439
- Chin J Vector Biol & Control, 2024, 35(4): 394-400, 439
- 10.11853/j.issn.1003.8280.2024.04.002
-
文章历史
- 收稿日期: 2024-01-29



2 贵州省疾病预防控制中心病媒生物监测科, 贵州省微生物组与传染性疾病防控重点实验室, 贵州 贵阳 550004;
3 遵义市疾病预防控制中心病媒生物防制科, 贵州 遵义 563000;
4 道真县疾病预防控制中心, 贵州 道真 563500
2 Vector Surveillance Section of Guizhou Center for Disease Control and Prevention, Key Laboratory of Microbio and Infectious Disease Prevention & Control in Guizhou Province, Guiyang, Guizhou 550004, China;
3 Vector Control Department of Zunyi Center for Disease Control and Prevention, Zunyi, Guizhou 563000, China;
4 Daozhen Center for Disease Control and Prevention, Daozhen, Guizhou 563500, China
蜱是重要的医学传播媒介,其生活史分为卵、幼蜱、若蜱和成蜱4个阶段,后3个阶段均需寄生在动物体表,通过吸食动物血液生存,期间可更换不同宿主并携带和传播多种病原体[1],是仅次于蚊的第二大病媒生物。蜱可携带约83种病毒、32种原虫、17种回归热螺旋体、14种细菌、2种线虫、1种衣原体、1种支原体、1种巴尔通体[2],可引起莱姆病、发热伴血小板减少综合征、人粒细胞无形体病等多种疾病。据估计,仅微小扇头蜱(Rhipicephalus microplus)在整个热带和亚热带地区每年可造成25亿美元的损失[3]。蜱传病原体研究具有重要的医学、兽医学和经济学意义。
由于广泛接触自然环境和多次更换宿主,蜱会携带多种病原菌、潜在致病菌和非致病菌,这些微生物群会对蜱的发育、繁殖、免疫以及宿主等产生影响。近年来气候变化产生了一些有利于蜱栖息地的条件,在中国检出的103种蜱传播的病原体中有65种是近20年来新发现的[4]。抗生素抗性基因(antibiotics resistance genes,ARGs)是一种新型污染物,在自然界广泛存在,对人类健康有很大的潜在风险。蜱是专性吸血的寄生虫,传播疾病的同时也传播ARGs,增加了相关蜱传疾病的治疗难度。因此,开展蜱微生物群和ARGs的研究对于了解蜱传疾病致病机制以及指导合理用药等有重要意义。
宏基因组学是一种直接提取微生物总核酸进行测序的方法,其可识别蜱体内大量病原体,并且不依赖于已知的核酸序列,是监测新出现的蜱传疾病的理想工具。贵州省地处我国西南,境内森林资源丰富、畜牧业发达,微小扇头蜱是该地区的优势蜱种之一[5],然而目前贵州省微小扇头蜱的微生物多样性情况尚未查清,人畜危害不明。因此,本研究采用宏基因组测序方法,对贵州省部分地区牛体表微小扇头蜱进行微生物群落结构和ARGs等的分析,旨在评估贵州省微小扇头蜱的人畜危害,以达到预测预警和有效防治蜱传疾病的目的。
1 材料与方法 1.1 样品采集根据前期调查基础,选择贵州省遵义市道真县旧城镇、安顺市紫云县火花镇、贵阳市修文县六广镇和六盘水市盘州市乌蒙镇4个蜱活动频繁地区,于2022-2023年每年的4月和5月在牛体表使用动物体表检蜱法采集蜱,置于-80℃保存。
1.2 形态学鉴定根据蜱的假头基形状、须肢、口下板齿式、盾板形状、气门板、足基节、爪垫等形态学特征,按照分类检索表鉴定微小扇头蜱。
1.3 DNA提取每个地区随机选取15只雌蜱,分为3管(道真、紫云和修文县以及盘州市分别编号为ZYWC、ASWC、GYWC和LPSWC),每管5只混样,分别用75%乙醇溶液和磷酸盐缓冲液(phosphate buffered saline,PBS)将蜱清洗3次以去除蜱表面污染,并放置到滤纸上吸干水分,随后加入液氮将蜱研磨成粉末状,按照E.Z.N.ATM Mag-Bind Soil DNA Kit(Omega Bio-Tek,美国)说明书提取蜱总DNA。
1.4 文库构建及测序提取的蜱DNA经1%琼脂糖凝胶电泳检测完整性,并使用Qubit 4.0荧光定量仪(Q33226,ThermoFisher)和Qubit™ dsDNA HS Assay Kit(ThermoFisher)对基因组浓度精确定量。使用Covaris超声波破碎仪(S220,Covaris)将检测合格的DNA裂解成约500 bp的片段,按照Hieff NGS® MaxUp Ⅱ DNA Library Prep Kit for Illumina®(上海翌圣生物科技有限公司)说明书进行文库构建,并用Hieff NGSTM DNA Selection Beads(上海翌圣生物科技有限公司)纯化扩增产物。使用Qubit 4.0荧光定量仪进行文库质控,文库的双端测序在DNBseq-T7测序平台(华大智造科技有限公司)上进行,测序策略为PE150。
1.5 数据质控使用Fastp 0.36软件按以下步骤对测序数据进行质量控制:①去除接头序列;②采用划窗法去除读长的3'~5'方向的低质量碱基,即去除 < 平均质量值Q20的碱基,划窗大小为4 bp;③剔除低质量序列,包括含大量模糊碱基序列(1条读长中有40%的碱基质量 < Q15)以及长度 < 35 nt的双端读长;④使用bowtie2 2.1.0将读长比对到蜱和牛基因组上,去除与蜱基因组和牛基因组相似性高的读长。
1.6 拼接组装与分箱首先使用megahit 1.2.9软件进行多样本混合拼接,得到初步的剪接重叠群(contig)序列。然后使用Bowite2 2.1.0清理读长,将其映射回剪接结果,提取未映射的读长,并使用SPAdes 3.13再次拼接以获得低丰度重叠群。使用Metawrap 1.3.2软件组合套件,依次完成Bin分选,Bin提纯,Bin定量,Bin重组装和Bin鉴定等过程。最终经过滤后得到完整度较高且低污染度的单菌基因组草图。
1.7 基因预测和非冗余基因集构建采用Prodigal 2.60软件对拼接结果进行开放阅读框(open reading frame,ORF)预测,选择长度≥100 bp的基因,将其翻译成氨基酸序列。对于各样本的基因预测结果,采用CD-HIT 2.60软件进行去冗余,获得非冗余的基因集。采用Bowtie2 2.1.0将质控后的读长比对到非冗余基因集序列上,利用SAMtools 1.5.0软件获得比对上的读长,并根据基因长度信息计算基因丰度。
1.8 物种和抗性基因注释将基因集与美国国立生物技术信息中心(National Center for Biotechnology Information,NCBI)非冗余蛋白库(Nr)和抗性基因数据库(structured ARG reference database,SARG)比对,获得基因的物种和抗性基因注释信息。基于基因集丰度信息和注释信息,得到物种和抗性基因的丰度信息,进行物种和抗性基因的组成和差异等的分析。
1.9 统计分析及绘图利用R 3.6.3软件进行组间相似性分析(analysis of similarities,ANOSIM)判断分组是否合理,P < 0.05为差异有统计学意义,并使用GraPhlAn 1.1.3软件绘制物种层级注释图。
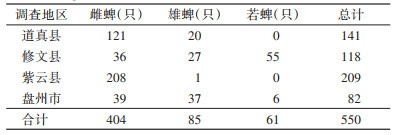
2 结果 2.1 形态学结果4个地区牛体表共采集到550只微小扇头蜱,均为吸血蜱,其中雌蜱404只,雄蜱85只,若蜱61只,其中道真、修文和紫云县以及盘州市分别采集到微小扇头蜱141、118、209和82只。见表 1。
|
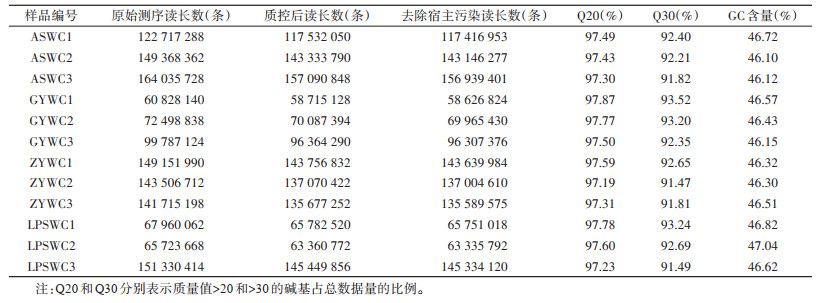
本次测序共获得1 388 623 524条原始读长,质控过滤后数据中测序错误率 < 1.00%(Q20)和 < 0.10%(Q30)的碱基数目比例分别达到97.19%和91.47%以上(表 2)。基因ORF预测共得到12 932 353个基因用于后续物种和功能注释,N50为357 bp,其中基因的平均长度为286.55 bp,最长的基因为25 875 bp。
|
将测序结果与NCBI-Nr数据库比对,共注释到15个门类,其中最丰富的是变形菌门(Proteobacteria),平均相对丰度为60.01%,其次是厚壁菌门(Firmicutes,36.76%)和放线菌门(Actinomycetota,2.98%),见图 1A、1B。在4个地区12组样品中,紫云和修文县的优势菌门为变形菌门和厚壁菌门,其中紫云县的平均相对丰度分别为45.40%、49.95%,修文县的平均相对丰度分别为49.14%、46.33%,道真县和盘州市的优势菌门为变形菌门,平均相对丰度分别为66.99%、78.49%。

|
| 注:ZYWC、ASWC、GYWC、LPSWC分别表示道真、紫云、修文县和盘州市的微小扇头蜱。 图 1 2022-2023年贵州省4个地区微小扇头蜱微生物群落结构 Figure 1 Microbial community structure of Rhipicephalus microplus in four regions of Guizhou Province from 2022 to 2023 |
| |
在属水平上,共注释到88个微生物属,丰度前10的菌属分别是克雷伯菌属(Klebsiella,38.58%)、葡萄球菌属(Staphylococcus,22.00%)、链球菌属(Streptococcus,11.21%)、立克次体属(Rickettsia,9.37%)、无形体属(Anaplasma,3.08%)、分枝杆菌属(Mycobacterium,1.77%)、柯克斯体属(Coxiella,1.25%)、肠球菌属(Enterococcus,1.16%)、硝酸盐还原菌属(Nitratireductor,1.14%)和Sinisalibacter(1.08%),见图 1C。4个地区样本中优势菌属均为克雷伯菌属,紫云、修文、道真县和盘州市的平均相对丰度分别为30.77%、32.21%、40.40%和50.95%。道真县和盘州市的微小扇头蜱立克次体属平均相对丰度较高,分别为16.30%和16.49%。
在种水平上,共注释到163个种,丰度前10的菌种分别是肺炎克雷伯菌(K. pneumoniae,38.58%)、金黄色葡萄球菌(Sta. aureus,21.90%)、肺炎链球菌(Str. pneumoniae,6.26%)、嗜吞噬细胞无形体(A. phagocytophilum,3.08%)、福尼尔立克次体(R. fournieri,3.04%)、咽峡炎链球菌(Str. anginosus,2.79%)、未分类立克次体属种类(unclassified Rickettsia species,2.46%)、变形链球菌(Str. mutans,2.07%)、结核分枝杆菌(M. tuberculosis,1.77%)、屎肠球菌(E. faecium,1.16%),见图 1D。其中紫云、修文和道真县,盘州市的肺炎克雷伯菌平均相对丰度分别为30.77%、32.21%、40.40%和50.95%,金黄色葡萄球菌平均相对丰度分别为29.52%、27.73%、18.30%和12.05%。本次调查检出了荆心立克次体(Candidatus R. jingxinensis,0.01%)、康氏立克次体(R. conorii,0.76%)、日本立克次体(R. japonica,0.70%)、西伯利亚立克次体(R. sibirica,0.06%)、莫纳森立克次体(R. monacensis,0.01%)等斑点热群立克次体(spotted fever group rickettsiae,SFGR),普氏立克次体(R. prowazekii,0.01%)和地方性斑疹伤寒立克次体(R. typhi,0.01%)等斑疹伤寒组立克次体,以及伽氏疏螺旋体(Borrelia garinii,0.01%)和贝氏柯克斯体(Coxiella burnetii,0.01%)等蜱传病原体。组间相似性分析结果显示组间差异大于组内差异(R=0.586,P=0.002)。
2.4 京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)功能注释将非冗余基因集蛋白序列与KEGG进行比对,结果显示4个地区的微小扇头蜱注释到基因数最多的通路是新陈代谢通路,基因数为13 270个,其次分别是人类疾病、遗传信息处理、环境信息处理、细胞过程和有机体系统,基因数分别为10 785、3 377、2 110、1 663和1 505个,见图 2 A。在与人类疾病相关通路注释到的基因中7 085个基因与传染病有关,包括细菌性传染病、病毒性传染病和寄生虫病,基因数分别是1 084、5 952和49个,1 957个基因与癌症通路有关,874个基因与心血管疾病通路有关,434个基因与耐药性通路有关,146个基因与神经退行性疾病有关,129个基因与内分泌和代谢疾病有关,86个基因与免疫性疾病有关。

|
| 注:ZYWC、ASWC、GYWC、LPSWC分别表示道真、紫云和修文县,盘州市的微小扇头蜱。 图 2 2022-2023年贵州省4个地区微小扇头蜱KEGG通路和抗生素抗性基因注释图 Figure 2 KEGG pathway and antibiotic resistance gene annotation of Rhipicephalus microplus in four regions of Guizhou Province from 2022 to 2023 |
| |
通过与SARG比对,4个地区微小扇头蜱共注释到3 316个ARGs,分别对β-内酰胺类(54.22%)、氨基糖苷类(20.18%)、氯霉素类(13.61%)和多药类(11.99%)抗生素产生抗药性。紫云和道真县的ARGs以β-内酰胺类为主,抗性基因数分别是595和1 301个,其中紫云县的β-内酰胺类ARGs包括TEM-1、TEM-157、TEM-171和TEM-197,道真县的β-内酰胺类ARGs包括TEM-1、TEM-102、TEM-168、TEM-171和TEM-197;盘州市的ARGs中氨基糖苷类和氯霉素类较多,分别是626和422个基因,氨基糖苷类ARGs包括aac(6')-I和ant(2")-I,氯霉素类ARGs包括氯霉素乙酰转移酶、氯霉素输出基因和cmlA,其次是多药类抗药相关基因,有294个,包括多药转运蛋白和adeJ;修文县的微小扇头蜱未注释到ARGs,见图 2B。为使数据更真实反映菌株ARGs丰度变化差异,采用基于16S rRNA拷贝数的方法对数据进行标准化转换,结果显示紫云和道真县的β-内酰胺类ARGs相对丰度分别为0.006%和0.004%,盘州市的氨基糖苷类、氯霉素类和多药类ARGs相对丰度分别为0.006%、0.004%和0.003%。见图 2 C。
3 讨论蜱是专性吸血的体外寄生虫,其体内微生物群组成复杂,培养法或者特异性引物PCR法等均无法全面了解蜱的微生物群落结构。本研究采用宏基因组测序方法对贵州省4个地区牛体表微小扇头蜱体内微生物群落、ARGs等开展研究,结果发现微小扇头蜱体内微生物群落结构相似,在门水平上均以变形菌门、厚壁菌门为主,该结果与Thanchomnang等[6]的研究结果相似。在属水平上,均以克雷伯菌属、葡萄球菌属为主,此外还有立克次体属、无形体属和柯克斯体属等,其所导致的立克次体病、无形体病和Q热等疾病由于缺乏典型的临床表现,极易漏诊和误诊,在人类社会中长期处于被低估的状态[7]。本研究中不同地区的立克次体属丰度不同,可能原因是蜱体内微生物群受到各地区地理环境、宿主的特异性、采样季节等的影响[8]。
在种水平上,4个地区的微小扇头蜱均以肺炎克雷伯菌和金黄色葡萄球菌为主,二者是常见的条件致病菌。丰度较高的还有嗜吞噬细胞无形体、福尼尔立克次体等致病菌,二者分别是人粒细胞无形体病和斑点热的病原体。20世纪90年代初,在美国首次发现了人粒细胞无形体病的确诊病例,随后我国于2006年首次在安徽省确诊了该病例,目前我国人粒细胞无形体病的病例报告主要发生在中部和中东部地区的湖北省和山东省,同时在我国东北部和东南部地区也发现了该病病例,然而贵州省尚未有此病例报道[4]。福尼尔立克次体隶属于SFGR,其于2013年在澳大利亚的仙燕锐缘蜱(Argas lagenoplastis)中首次检出[9]。2023年向昱龙等[10]在贵州省少数民族地区采集的微小扇头蜱中首次在中国报道了福尼尔立克次体,本研究在贵州省其他地区微小扇头蜱中检出该病原体,提示贵州省应加强对福尼尔立克次体的研究,了解其分布情况和致病机制。
本研究检出了多种SFGR的基因型,其中日本立克次体、西伯利亚立克次体、莫纳森立克次体是中国已证实的对人类致病的8种SFGR之一[11]。康氏立克次体是地中海斑点热的病原体,主要流行于地中海盆地,其传播媒介是血红扇头蜱(R. sanguineus)[12],目前并未在中国发现该病例,此次在微小扇头蜱体内检测到该病原体,提示微小扇头蜱可能也是其传播媒介。近年来在中国安徽、山东省等地也报道了日本立克次体的存在[13-14]。此前研究者在贵州省安顺市[15]、黔东南州[16]、黔西南州、六盘水市和毕节市[17]等地的蜱中也检出了荆心立克次体,提示贵州省很可能也是蜱媒立克次体病的自然疫源地,应加强对蜱媒立克次体的监测与控制,从而预防蜱传立克次体病的发生。本研究还检出了斑疹伤寒组立克次体、伽氏疏螺旋体和贝氏柯克斯体等病原体,可分别导致斑疹伤寒、莱姆病和Q热,斑疹伤寒在我国属于法定丙类传染病[18],此前研究者也曾在贵州省发现莱姆病和Q热感染病例[19],提示微小扇头蜱可能是几种疾病的传播媒介之一,应当引起广泛的重视。
KEGG功能注释发现4个地区均是新陈代谢相关通路的基因数最多,可能原因是蜱的生活史复杂[20],且雌蜱在吸血后体形往往膨大数倍,为保证良好的生长发育,需要较强的新陈代谢能力。在与人类疾病相关通路中,与传染病和寄生虫病相关的基因数最多,同时还有部分与耐药性相关的基因,这可能与蜱本身能够携带和传播众多病原体和ARGs有关[21]。蜱中病原体除可引起多种人畜共患病外,还可作为潜在的抗性基因贮藏库[22],通过血餐向人类和动物传播抗性基因。研究注释到与β-内酰胺类、氨基糖苷类、氯霉素类和多药类抗生素相关的4类抗性基因,不同地区抗性基因的种类和相对丰度差异较大,这与Wei等[23]的研究结果相似。研究表明动物制品和城市污水中含有多种抗性基因[24],由于蜱是以吸血为生的体外寄生虫,因此本研究中发现的抗性基因极有可能来源于微小扇头蜱的寄生宿主[25]。同时抗性基因也可随禽畜粪便等进入外界环境[26],污染水体和土壤等,从而影响人类食品,极大地危害人类健康。本研究评估了贵州省微小扇头蜱中ARGs的多样性,强调了其作为ARGs的传播者进入环境和脊椎动物宿主的可能作用,然而不同地区的抗生素使用惯例是否是主要因素需要进一步研究。因此,蜱及其宿主体内ARGs的种类分布和数量动态应当引起重视。
本研究采用宏基因组测序方法,对贵州省紫云、道真和修文县以及盘州市牛体表微小扇头蜱进行微生物群落多样性分析、KEGG功能注释和ARGs注释,结果发现蜱体内微生物菌群和抗生素抗性基因组成丰富,与人类疾病相关通路的基因较多,不同地区微小扇头蜱的菌属相对丰度和抗性基因的种类和丰度不同,在种水平上,蜱体内既有SFGR、斑疹伤寒立克次体、贝氏柯克斯体等致病菌,又有肺炎克雷伯菌、金黄色葡萄球菌等丰度较高但是否经蜱媒致病尚不明确的菌种,提示贵州省应加强对蜱微生物群的研究以了解蜱传疾病的致病机制,研究结果对于贵州省指导合理用药、预防蜱传疾病等有重要意义。同时,贵州省生境多样、蜱种丰富,本研究涉及的采样点和蜱种较少,后续将进一步增加采样点、样本的种类和数量,以全面了解贵州省的蜱种类分布及微生物携带情况,为蜱媒疾病的有效防控提供参考依据。
志谢 本次调查得到遵义市、道真县、紫云县、修文县和盘州市疾病预防控制中心的大力支持和帮助,特此一并表示感谢利益冲突 无
| [1] |
曹国平, 占炳东, 钟建跃, 等. 2017-2019年浙江省衢州市城区公园蜱分布及携带病原体情况调查[J]. 疾病监测, 2021, 36(9): 879-883. Cao GP, Zhan BD, Zhong JY, et al. Status of tick distribution and tick-borne pathogens in urban parks of Quzhou, Zhejiang, 2017-2019[J]. Dis Surveill, 2021, 36(9): 879-883. DOI:10.3784/jbjc.202106010314 |
| [2] |
Beard D, Stannard HJ, Old JM. Parasites of wombats (family Vombatidae), with a focus on ticks and tick-borne pathogens[J]. Parasitol Res, 2021, 120(2): 395-409. DOI:10.1007/s00436-020-07036-0 |
| [3] |
Barker SC, Walker AR. Ticks of Australia. The species that infest domestic animals and humans[J]. Zootaxa, 2014, 3816(1): 1-144. DOI:10.11646/zootaxa.3816.1.1 |
| [4] |
Zhao GP, Wang YX, Fan ZW, et al. Mapping ticks and tick-borne pathogens in China[J]. Nat Commun, 2021, 12(1): 1075. DOI:10.1038/s41467-021-21375-1 |
| [5] |
Xiang YL, Zhou JZ, Yu FX, et al. Characterization of bacterial communities in ticks parasitizing cattle in a touristic location in southwestern China[J]. Exp Appl Acarol, 2023, 90(1/2): 119-135. DOI:10.1007/s10493-023-00799-y |
| [6] |
Thanchomnang T, Rodpai R, Thinnabut K, et al. Characterization of the bacterial microbiota of cattle ticks in northeastern Thailand through 16S rRNA amplicon sequencing[J]. Infect Genet Evol, 2023, 115: 105511. DOI:10.1016/j.meegid.2023.105511 |
| [7] |
Lu M, Tang GP, Ren ZQ, et al. Ehrlichia, Coxiella and Bartonella infections in rodents from Guizhou Province, southwest China[J]. Ticks Tick Borne Dis, 2022, 13(5): 101974. DOI:10.1016/j.ttbdis.2022.101974 |
| [8] |
Jia N, Wang JF, Shi WQ, et al. Large-scale comparative analyses of tick genomes elucidate their genetic diversity and vector capacities[J]. Cell, 2020, 182(5): 1328-1340.e13. DOI:10.1016/j.cell.2020.07.023 |
| [9] |
Diop A, Barker SC, Eberhard M, et al. Rickettsia fournieri sp. nov., a novel spotted fever group Rickettsia from Argas lagenoplastis ticks in Australia[J]. Int J Syst Evol Microbiol, 2018, 68(12): 3781-3784. DOI:10.1099/ijsem.0.003057 |
| [10] |
向昱龙, 周敬祝, 张燕, 等. 贵州省少数民族自治州微小扇头蜱的宏基因组分析[J]. 中国媒介生物学及控制杂志, 2023, 34(3): 319-325. Xiang YL, Zhou JZ, Zhang Y, et al. Metagenomic analysis of Rhipicephalus microplus from minority autonomous prefectures in Guizhou Province, China[J]. Chin J Vector Biol Control, 2023, 34(3): 319-325. DOI:10.11853/j.issn.1003.8280.2023.03.007 |
| [11] |
韩婧, 贺真, 邵中军. 常见蜱传立克次体的研究进展[J]. 中华卫生杀虫药械, 2022, 28(1): 86-89. Han J, He Z, Shao ZJ. The research progress of common tick-borne Rickettsia[J]. Chin J Hyg Insect Equip, 2022, 28(1): 86-89. DOI:10.19821/j.1671-2781.2022.01.024 |
| [12] |
Spernovasilis N, Markaki I, Papadakis M, et al. Mediterranean spotted fever: Current knowledge and recent advances[J]. Trop Med Infect Dis, 2021, 6(4): 172. DOI:10.3390/tropicalmed6040172 |
| [13] |
Li JB, Hu W, Wu T, et al. Japanese spotted fever in eastern China, 2013[J]. Emerg Infect Dis, 2018, 24(11): 2107-2109. DOI:10.3201/eid2411.170264 |
| [14] |
Qin XR, Han HJ, Han FJ, et al. Rickettsia japonica and novel Rickettsia species in ticks, China[J]. Emerg Infect Dis, 2019, 25(5): 992-995. DOI:10.3201/eid2505.171745 |
| [15] |
Wang Q, Guo WB, Pan YS, et al. Detection of novel spotted fever group rickettsiae (Rickettsiales: Rickettsiaceae) in ticks (Acari: Ixodidae) in southwestern China[J]. J Med Entomol, 2021, 58(3): 1363-1369. DOI:10.1093/jme/tjaa294 |
| [16] |
Lu M, Meng C, Zhang B, et al. Prevalence of spotted fever group Rickettsia and Candidatus Lariskella in multiple tick species from Guizhou province, China[J]. Biomolecules, 2022, 12(11): 1701. DOI:10.3390/biom12111701 |
| [17] |
Lu M, Meng C, Gao X, et al. Diversity of Rickettsiales in Rhipicephalus microplus ticks collected in domestic ruminants in Guizhou Province, China[J]. Pathogens, 2022, 11(10): 1108. DOI:10.3390/pathogens11101108 |
| [18] |
包蕾, 李园园, 李海艳, 等. 云南省西双版纳州2016-2020年斑疹伤寒流行特征及影响因素研究[J]. 中国媒介生物学及控制杂志, 2022, 33(6): 854-858. Bao L, Li YY, Li HY, et al. Epidemiological characteristics and influencing factors of typhus in Xishuangbanna Dai Autonomous Prefecture, Yunnan Province, China, 2016-2020[J]. Chin J Vector Biol Control, 2022, 33(6): 854-858. DOI:10.11853/j.issn.1003.8280.2022.06.017 |
| [19] |
Wu XB, Na RH, Wei SS, et al. Distribution of tick-borne diseases in China[J]. Parasit Vectors, 2013, 6(1): 119. DOI:10.1186/1756-3305-6-119 |
| [20] |
Estrada-Peña A, De La Fuente J. The ecology of ticks and epidemiology of tick-borne viral diseases[J]. Antiviral Res, 2014, 108: 104-128. DOI:10.1016/j.antiviral.2014.05.016 |
| [21] |
Li SS, Zhang XY, Zhou XJ, et al. Bacterial microbiota analysis demonstrates that ticks can acquire bacteria from habitat and host blood meal[J]. Exp Appl Acarol, 2022, 87(1): 81-95. DOI:10.1007/s10493-022-00714-x |
| [22] |
许雪莲, 韩阿祥, 叶诗晴, 等. 温州口岸截获蜱体内微生物群落结构、抗生素抗性基因及毒力因子的宏基因组分析[J]. 中国媒介生物学及控制杂志, 2021, 32(6): 763-771. Xue XL, Han AX, Ye SQ, et al. Metagenomic analysis of microbial community structure, antibiotic resistance genes, and virulence factors of ticks captured at Wenzhou port, Zhejiang Province, China[J]. Chin J Vector Biol Control, 2021, 32(6): 763-771. DOI:10.11853/j.issn.1003.8280.2021.06.021 |
| [23] |
Wei NN, Lu JM, Dong Y, et al. Profiles of microbial community and antibiotic resistome in wild tick species[J]. mSystems, 2022, 7(4): e0003722. DOI:10.1128/msystems.00037-22 |
| [24] |
Chavarría-Bencomo IV, Nevárez-Moorillón GV, Espino-Solís GP, et al. Antibiotic resistance in tick-borne bacteria: A one health approach perspective[J]. J Infect Public Health, 2023, 16 Suppl 1: 153-162. DOI:10.1016/j.jiph.2023.10.027 |
| [25] |
Papp M, Tóth AG, Valcz G, et al. Antimicrobial resistance gene lack in tick-borne pathogenic bacteria[J]. Sci Rep, 2023, 13(1): 8167. DOI:10.1038/s41598-023-35356-5 |
| [26] |
Zhang WM, Yu CX, Yin SQ, et al. Transmission and retention of antibiotic resistance genes (ARGs) in chicken and sheep manure composting[J]. Bioresour Technol, 2023, 382: 129190. DOI:10.1016/j.biortech.2023.129190 |