 2023, Vol. 34
2023, Vol. 34扩展功能
文章信息
- 樊洁丽, 刘焱晖, 殷雅楠, 赵建国, 孙定炜, 廖承红, 韩谦
- FAN Jie-li, LIU Yan-hui, YIN Ya-nan, ZHAO Jian-guo, SUN Ding-wei, LIAO Cheng-hong, HAN Qian
- 基于宏基因组学分析海南省不同养殖场库蠓共生微生物群落组成
- A metagenomic analysis of the composition of symbiotic microbial communities in Culicoides on different livestock farms in Hainan Province
- 中国媒介生物学及控制杂志, 2023, 34(4): 472-479
- Chin J Vector Biol & Control, 2023, 34(4): 472-479
- 10.11853/j.issn.1003.8280.2023.04.006
-
文章历史
- 收稿日期: 2023-02-06



2 海南省疾病预防控制中心, 海南 海口 570203
2 Hainan Center for Disease Control and Prevention, Haikou, Hainan 570203, China
蠓科(Ceratopogonidae)属于双翅目的小型昆虫,包括库蠓属(Culicoides)、细蠓属(Leptconops)、蠛蠓属(Lasiohelea)和澳蠓属(Austroconops)4属吸血蠓,库蠓属是蠓科中最大的属,也是吸血蠓中与人畜关系最密切、分布最广、种类最多的属[1]。库蠓作为媒介昆虫,通过刺吸人和动物血液可传播多种病原体,如土拉弗氏杆菌(Bacterium tularensis)[2]、住白细胞原虫(Leucocytozoon)[3]、蓝舌病病毒(Bluetongue virus)[4]、施马伦贝格病毒(Schmallenberg virus)[5]、流行性出血热病毒(Epizootic hemorrhagic disease virus)[6]等。海南岛地处热带,年平均气温较高,雨量充沛,植被茂盛,为多种病媒生物提供了良好的孳生栖息环境。有研究对海南省5市(县)禽畜厩舍的吸血蠓进行调查,发现不同市(县)、不同场所的吸血蠓优势种组成不同[7]。叶雅芳等[8]利用形态学与分子生物学方法相结合对104只库蠓进行鉴定,刘帅等[9]从新疆维吾尔自治区(新疆)搜集的库蠓中分离并鉴定出盖塔病毒(Getah virus),Duan等[10]从云南省采集的连斑库蠓(C. jacobsoni)中分离出西藏环状病毒(Tibet orbivirus)。目前对库蠓的研究主要集中在物种鉴定、病原分离与检测方面,而对库蠓共生微生物方面的研究则少有报道。
昆虫体内共生微生物种类丰富,包括细菌、真菌、古菌、病毒以及一些小型原生生物。共生微生物在宿主的各项生命活动中发挥着重要的调节作用,包括消化食物、供给重要营养、提高宿主的防御和解毒能力、影响发育周期、寿命、交配与繁殖等[11-12]。昆虫共生微生物是通常由环境、食物中获得,被肠道环境选择的微生物,因此肠道微生物的组成与食物种类和肠道环境密切相关[13]。微生物与昆虫宿主复杂的关系成为科学研究的焦点之一,对共生微生物的深入研究有利于开发生物杀虫剂,为未来针对害虫的生物防治提供新策略。
1 材料与方法 1.1 库蠓采集2021年8-9月,选取海南省东部文昌市、中部定安县和南部乐东县共5个不同的养殖场(牛圈、鸡舍、犬场、羊圈、猪圈)作为采样点,采用紫外灯诱法在傍晚至次日清晨采集库蠓。通过形态学和分子生物学方法对库蠓进行分类,存于95% 乙醇溶液,-20 ℃备用。
1.2 宏基因组测序分别使用95%乙醇溶液和ddH2O对库蠓做涡旋清洗,以去除其表面携带的微生物,将清洗好的蠓置于超净台风干水分,每个采样养殖场各称取10 mg库蠓(约100只)作为一组,使用十六烷基三甲基溴化铵法提取总DNA,利用NanoDrop 2000检测DNA纯度,1%琼脂糖凝胶电泳检测基因组DNA完整性。质检合格的基因组DNA由广州基迪奥生物公司构建宏基因组文库和上机测序。
1.3 生物信息学分析用FASTP(v0.18.0)对Illumina平台的原始数据进行过滤得到高质量数据,将数据比对昆虫数据库以及库蠓属基因组,去除比对上的高相似度读长,以此剔除宿主基因组。使用MEGAHIT(v1.1.2)软件对高质量数据进行组装得到重叠序列。利用MetaGeneMark(v3.38)对 > 500 bp的重叠序列进行基因预测。选取所有长度≥300 bp的基因序列,使用CD-HIT(v4.6)去冗余获得非冗余基因集,参数设置为相似度≥95%且读长覆盖度 > 90%,并使用Bowtie(v2.2.5)最终获得非冗余基因集,通过DIAMOND(v0.9.24)与京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)数据库比对进行基因功能注释,使用Kaiju(v1.6.3)将读长比对非冗余蛋白质的氨基酸序列数据库进行物种注释。
2 结果 2.1 库蠓采集信息2021年8-9月在海南省乐东县佛罗镇青山村的牛圈、羊圈和猪圈,文昌市潭牛镇195县道的鸡舍以及定安县新竹镇大路村的犬场采样,牛圈、羊圈和猪圈的优势库蠓为尖喙库蠓(C. oxystoma),鸡舍和犬场的优势库蠓为荒川库蠓(C. arakawae)。见表 1。
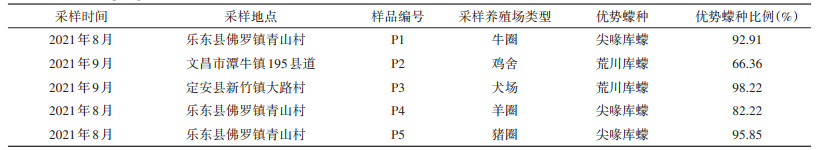
|
5个库蠓样本测序共获得原始数据54.96 G,质控后得到有效数据共54.60 G,质控后的有效数据率为99.34%,Q20值达97.11%。见表 2。
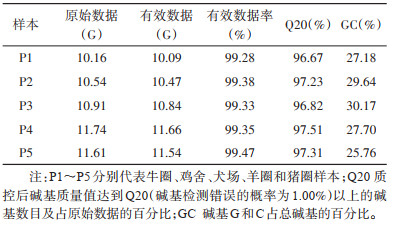
|
从门水平上,根据物种分类丰度用物种堆叠图展示总丰度排名前10的物种(图 1)。93.23%的物种可以注释到门水平,排名前10的门占76.46%,有变形菌门(Proteobacteria)、子囊菌门(Ascomycota)、厚壁菌门(Firmicutes)、担子菌门(Basidiomycota)、放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、毛霉菌门(Mucoromycota)、壶菌门(Chytridiomycota)、绿藻门(Chlorophyta)和捕虫霉门(Zoopagomycota),分别占33.78%、9.09%、8.88%、5.36%、5.18%、3.86%、3.60%、2.28%、2.23%和2.18%。变形菌门的相对丰度最高,在P1~P5的比例分别是28.38%、42.32%、31.48%、23.29%和43.45%,其中在P4占比最少,在P5占比最多;子囊菌门占比分别为9.58%、7.95%、10.16%、10.39%和7.36%;厚壁菌门占比分别为13.35%、5.69%、5.24%、13.08%和7.06%,在P1和P4占比较高且比例接近;其余门占比均 < 6.00%。

|
| 注:P1~P5分别代表牛圈、鸡舍、犬场、羊圈和猪圈样本;不同颜色代表门水平不同的物种,堆叠图展示优势门样本占比变化趋势。 图 1 2021年海南省3个市(县)5个不同养殖场库蠓门水平上的微生物群落结构 Figure 1 Microbial community structures of Culicoides at the phylum level on 5 different livestock farms in 3 cities (counties) in Hainan Province, 2021 |
| |
在属水平上,相对含量在前15的属,丰度由高到低分别是不动杆菌属(Acinetobacter,22.48%)、梭菌属(Clostridium,7.25%)、柠檬酸杆菌属(Citrobacter,5.42%)、沃尔巴克氏体属(Wolbachia,5.30%)、管鞭毛虫属(Salpingoeca,5.25%)、单领藻属(Monosiga,4.89%)、乳酸杆菌属(Lactobacillus,3.29%)、假单胞菌属(Pseudomonas,3.37%)、气单胞菌属(Aeromonas,3.24%)、埃希菌属(Escherichia,3.13%)、肠杆菌属(Enterobacter,2.91%)、锥虫属(Trypanosoma,2.80%)、疟原虫属(Plasmodium,2.44%)、曲霉属(Aspergillus,2.31%)和乳球菌属(Lactococcus,2.17%)(图 2)。其中不动杆菌属作为第一大优势属,其在P1~P5中的占比分别为5.44%、27.70%、36.30%、3.93%和39.05%,在P4占比为5个养殖场最低,在P2、P3和P5占比较高;梭菌属在5个样本中的比例为16.15%、1.35%、1.27%、16.29%和1.21%,与不动杆菌属在各样本中的占比情况刚好相反;占比 < 10.00%的属中,柠檬酸杆菌属、沃尔巴克氏体属和乳酸杆菌属在样本中的占比情况均是在P1、P4和P5占比较在P2和P3高,但占比相似,而管鞭毛虫属、单领藻属、假单胞菌属、埃希菌属、肠杆菌属、锥虫属和曲霉属的情况均是在P5占比最低,在剩余样本占比相似。

|
| 注:P1~P5分别代表牛圈、鸡舍、犬场、羊圈和猪圈样本;图形最外环左半圆颜色表示各属,右半圆颜色表示各样本;内圈坐标刻度表示对应颜色属的占比(%);内环的弦由属发出,指向样本;弦粗细表示样本对应属的数值高低。 图 2 2021年海南省3个市(县)5个不同养殖场库蠓属水平上的微生物群落结构 Figure 2 Microbial community structures of Culicoides at the genus level on 5 different livestock farms in 3 cities (counties) in Hainan Province, 2021 |
| |
从基于属水平的物种upset plot图(图 3)可知,5个养殖场共有属为3 357属,属的种类丰富度排序为P3 > P2 > P4 > P1 > P5,P1~P4共有139属,P1~P5特有属数分别为49、113、192、43和49属。利用物种PCoA图(图 4)展示5个样本属水平的微生物群落结构差异,第1轴贡献度为49.50%,第2轴贡献度为38.49%,P1与P4空间距离最近,P2与P3空间距离较近,而P5与其他样本距离最远。

|
| 注:P1~P5分别代表牛圈、鸡舍、犬场、羊圈和猪圈样本;左侧水平柱子表示各样本拥有的属数量,图形下方矩阵中的单个点表示统计某个样本特有的元素,点和点之间的连线表示统计相连样本的交集,右上方竖直柱子表示统计的特有或交集的属数量。 图 3 2021年海南省3个市(县)5个不同养殖场库蠓基于属水平的微生物多样性分析 Figure 3 Microbial diversity analysis of Culicoides at the genus level on 5 different livestock farms in 3 cities (counties) in Hainan Province, 2021 |
| |

|
| 注:P1~P5分别代表牛圈、鸡舍、犬场、羊圈和猪圈样本;基于相异矩阵系数距离进行分析,以样本在二维平面的距离关系反映样本间菌群结构的相似性,样本距离越近,说明样本间属水平物种组成越相似;轴标签括号中的百分比表示各坐标轴占菌群结构总变异的解释度,平面两个轴的解释度越高,说明模型效果越好。 图 4 2021年海南省3个市(县)5个不同养殖场库蠓共生微生物基于属水平的主坐标分析 Figure 4 Principal coordinate analysis of the symbiotic microorganism in Culicoides at the genus level on 5 different livestock farms in 3 cities (counties) in Hainan Province, 2021 |
| |
5个不同养殖场样本共有基因数目1 065个,P1~P5独有的基因数目分别是7 825、35 061、50 486、31 219和11 469,P3独有基因数目最多。P1和P4的共有基因数最多,为31 350个,其次为P1、P4、P5共有17 546个,P2、P3共有15 458个(图 5A)。P1和P4的优势库蠓都是尖喙库蠓和原野库蠓(C. homotomus),P5的优势库蠓为尖喙库蠓,而P2和P3的优势库蠓则是荒川库蠓。为了进一步比较样本之间的相似性,基于基因丰度计算样品间的Pearson相关系数,从样品间关系热图可以看出,P1和P4颜色更深,2个采样地样品的基因丰度的模式相似度最高,其次是P2和P3(图 5B),与基因数目差异分析结果一致。

|
| 注:P1~P5分别代表牛圈、鸡舍、犬场、羊圈和猪圈样本;A库蠓基于基因数目的韦恩图,花瓣内数字代表不同样本共有或独有的基因数目;B库蠓基于基因数目的样品相关性热图,颜色表示样品的相关性,颜色越深,两样品基因相关性越强。 图 5 2021年海南省3个市(县)5个不同养殖场库蠓基因差异分析 Figure 5 Gene differential analysis of Culicoides on 5 different livestock farms in 3 cities (counties) in Hainan Province, 2021 |
| |
77 369(36.25%)个非冗余基因集与KEGG数据库比对分析。KEGG功能注释到的6个A级代谢通路中,新陈代谢中P5的相对丰度与其他样品差异明显,而人类疾病、生物体系统、遗传信息处理、细胞过程和环境信息处理5个代谢通路中P2和P3相对丰度较高且相似,而P1、P4和P5相对丰度较低(图 6)。从KEGG注释基因数目统计图(图 7)可知,新陈代谢通路注释到的数量最多,占总注释基因数目的29.10%,其次是人类疾病、生物体系统、细胞过程、遗传信息处理以及环境信息处理。其中微生物在新陈代谢中的功能主要是碳水化合物代谢、氨基酸代谢、脂质代谢、辅助因子和维生素代谢,注释到的基因数目分别是4 326、3 741、2 532和2 377个。而与人类疾病相关代谢通路中注释到与传染病通路相关3 702个、癌症通路相关2 019个、神经退行性疾病通路相关1 479个、耐药性通路相关1 402个。

|
| 注:P1~P5分别代表牛圈、鸡舍、犬场、羊圈和猪圈样本;横坐标为各样本,纵坐标为6个A级代谢通路的相对丰度;颜色深浅代表相对丰度大小。 图 6 2021年海南省3个市(县)5个不同养殖场库蠓功能基因注释热图 Figure 6 Heatmap of functional gene annotation of Culicoides on 5 different livestock farms in 3 cities (counties) in Hainan Province, 2021 |
| |

|
| 注:P1~P5分别代表牛圈、鸡舍、犬场、羊圈和猪圈样本;纵坐标表示B级水平代谢通路。 图 7 2021年海南省3个市(县)5个不同养殖场库蠓体内共生微生物代谢通路注释基因数目统计 Figure 7 Statistics of the number of annotated genes by metabolic pathways of the symbiotic microorganism in Culicoides on 5 different livestock farms in 3 cities (counties) in Hainan Province, 2021 |
| |
昆虫共生微生物与宿主的生命活动息息相关,库蠓体内共生微生物群落结构复杂,靠传统的分离培养无法全面了解其微生物群落。因此,本研究利用宏基因组测序技术探究海南省不同养殖场吸血库蠓的共生微生物群落组成及功能。
在海南省5个不同的养殖场中,主要优势物种分布不同,牛圈、羊圈和猪圈均以尖喙库蠓为优势种,而鸡舍和犬场优势物种为荒川库蠓。变形菌门作为库蠓体内共生微生物的第一大优势菌门,同样是双翅目的致倦库蚊(Culex pipiens quinquefasciatus)、鳞翅目的小菜蛾(Plutella xylostella)和等翅目黄翅大白蚁(Macrotermes barneyi)的优势菌门,其作为优势菌门在这些昆虫不同样本中具有较高的一致性[14-16]。继变形菌门之后的优势菌门中,厚壁菌门是肠道最主要的菌门之一,大部分细菌可以消化纤维、将纤维素分解为宿主可利用的挥发性脂肪酸,部分细菌可作为产热菌作用于食物发酵加速碳水化合物的氧化,以增加从食物中获取的能量,而拟杆菌门可帮助宿主降解非结构性碳水化合物和蛋白质[17-18]。在属水平上,5个样本的优势菌属有不动杆菌属、梭菌属、柠檬酸杆菌属和沃尔巴克氏体属等。其中,不动杆菌在自然界广泛分布,是医学临床感染中常见的条件致病菌之一,可引起肺炎、败血症、脑膜炎、外伤感染、尿路感染等[19]。伊蚊和按蚊中普遍存在不动杆菌,而库蠓的孳生栖息环境和食物来源与它们相似,这表明不同昆虫可以从相似的生活环境中获得一致的微生物[20]。沃尔巴克氏体属作为一种胞内共生菌广泛分布于昆虫中,有研究表明超65%的昆虫天然携带沃尔巴克氏体[21]。此外沃尔巴克氏体基本传播方式为母系传播,可以通过诱导孤雌生殖、雌性化、杀雄等调控宿主的生殖,而这也使得其能广泛寄生[22]。值得注意的是,寄生于宿主血液,可以引起人兽共患锥虫病的锥虫属也有被发现,而此前也有研究在滴斑库蠓(C. guttifer)中检测到锥虫[23]。锥虫科的寄生虫经常被发现存在昆虫体内,尤其是在双翅目与半翅目昆虫,而本实验在双翅目的库蠓中发现了锥虫属,推测库蠓可能是锥虫属的媒介之一,但仍需要进一步验证。更值得关注的是,由按蚊传播的疟原虫也被检测到,也就是说吸血库蠓有可能是疟原虫的潜在传播媒介,而证实重要寄生虫的宿主有助于疾病预防和控制。
不同的养殖场库蠓共生微生物优势菌群相同,在门水平上均以变形菌门、子囊菌门和厚壁菌门为主要菌门,在属水平上均以不动杆菌属和梭菌属为优势菌属,表明库蠓共生微生物的核心菌群相对稳定,推测养殖场对库蠓微生物群落主成分影响不大。但从属水平的微生物多样性分析显示,不同的养殖场库蠓微生物群落虽然大部分相同,但每个养殖场有其独特的微生物群落,这可能与库蠓种类和生活环境相关。库蠓种类组成和生存环境越相似,共生微生物群落越相似。基因数目和基因代谢通路差异分析结果也显示,牛圈、羊圈和猪圈养殖场和库蠓种类组成相似的3个样品差异更小。库蠓成虫雄性吸食植物汁液,雌性可刺吸人、畜血液传播病原微生物,所以不同的养殖场库蠓共生微生物群落的差异与其食物种类也有一定的关系。研究表明,微生物在营养物质的代谢过程起到了关键作用,微生物与宿主之间的联系主要体现在微生物食物链提供的直接底物[24]。依据KEGG数据统计,微生物代谢通路主要功能是新陈代谢,包括碳水化合物代谢、氨基酸代谢、脂质代谢、辅助因子和维生素代谢以及能量代谢等,这与成虫库蠓以植物汁液、人畜血液为食的食性相吻合。第二大代谢通路功能是人类疾病,包括传染病、癌症、神经退行性疾病、耐药性等,这与库蠓作为病媒生物传播多种病原体的特征相符。
库蠓赖以生存的环境中微生物群落组成、多样性、丰度及食物种类与来源等因素会直接影响其共生微生物群落组成和结构。本研究通过宏基因组测序技术对海南省不同养殖场中库蠓的共生微生物进行分析,发现库蠓共生微生物的核心菌群相对稳定,不同养殖场库蠓微生物多样性、基因数目与基因功能的差异与库蠓种类密切相关。本研究对于了解媒介生物库蠓共生微生物群落组成及其功能有重要意义,也为今后库蠓的生物防控提供新思路。
利益冲突 无
| [1] |
Borkent A, Dominiak P. Catalog of the biting midges of the world (Diptera: Ceratopogonidae)[J]. Zootaxa, 2020, 4787(1): 1-377. DOI:10.11646/zootaxa.4787.1.1 |
| [2] |
霍坚, 高永利, 郑龙, 等. 土拉弗朗西斯菌病的特征与防治[J]. 中华卫生杀虫药械, 2021, 27(2): 182-186. Huo J, Gao YL, Zheng L, et al. Research progress of characteristics, prevention and treatment on tularemia[J]. Chin J Hyg Insect Equip, 2021, 27(2): 182-186. DOI:10.19821/j.1671-2781.2021.02.023 |
| [3] |
Yu CY, Wang JS, Yeh CC. Culicoides arakawae (Diptera: Ceratopogonidae) population succession in relation to leucocytozoonosis prevalence on a chicken farm in Taiwan[J]. Vet Parasitol, 2000, 93(2): 113-120. DOI:10.1016/s0304-4017(00)00362-9 |
| [4] |
Duan YL, Li L, Bellis G, et al. Detection of Bluetongue virus in Culicoides spp. in southern Yunnan province, China[J]. Parasit Vectors, 2021, 14(1): 68. DOI:10.1186/s13071-020-04518-z |
| [5] |
Ségard A, Gardès L, Jacquier E, et al. Schmallenberg virus in Culicoides Latreille (Diptera: Ceratopogonidae) populations in France during 2011-2012 outbreak[J]. Transbound Emerg Dis, 2018, 65(1): e94-e103. DOI:10.1111/tbed.12686 |
| [6] |
Mcgregor BL, Shults PT, Mcdermott EG. A review of the vector status of North American Culicoides (Diptera: Ceratopogonidae) for Bluetongue virus, Epizootic hemorrhagic disease virus, and other arboviruses of concern[J]. Curr Trop Med Rep, 2022, 9(4): 130-139. DOI:10.1007/s40475-022-00263-8 |
| [7] |
刘国平, 孙定炜, 范娜, 等. 海南省5市县禽畜厩舍吸血蠓调查[J]. 中国热带医学, 2020, 20(5): 413-416, 428. Liu GP, Sun DW, Fan N, et al. Hematophagous midges at the hencoop and livestock sheds in five counties of Hainan[J]. China Trop Med, 2020, 20(5): 413-416, 428. DOI:10.13604/j.cnki.46-1064/r.2020.05.04 |
| [8] |
叶雅芳, 刘德星, 李婷婷, 等. 广东省中山市库蠓形态与分子鉴定[J]. 中国人兽共患病学报, 2019, 35(11): 1021-1028. Ye YF, Liu DX, Li TT, et al. Morphological and molecular identification of Culicoides collected in Zhongshan, Guangdong, China[J]. Chin J Zoonoses, 2019, 35(11): 1021-1028. DOI:10.3969/j.issn.1002-2694.2019.00.173 |
| [9] |
刘帅, 加帕尔·哈斯木, 薛新梅, 等. 新疆库蠓源性盖塔病毒的分离与鉴定[J]. 畜牧兽医学报, 2017, 48(10): 1998-2004. Liu S, Jiapaer HSM, Xue XM, et al. Isolation and identification of Getah virus from Culicoides in Xinjiang[J]. Acta Veterin Zootechn Sin, 2017, 48(10): 1998-2004. DOI:10.11843/j.issn.0366-6964.2017.10.024 |
| [10] |
Duan YL, Yang ZX, Bellis G, et al. Isolation of Tibet orbivirus from Culicoides jacobsoni (Diptera, Ceratopogonidae) in China[J]. Parasit Vectors, 2021, 14(1): 432. DOI:10.1186/s13071-021-04899-9 |
| [11] |
Storelli G, Defaye A, Erkosar B, et al. Lactobacillus plantarum promotes Drosophila systemic growth by modulating hormonal signals through TOR-dependent nutrient sensing[J]. Cell Metab, 2011, 14(3): 403-414. DOI:10.1016/j.cmet.2011.07.012 |
| [12] |
Cirstea MS, Yu AC, Golz E, et al. Microbiota composition and metabolism are associated with gut function in Parkinson's disease[J]. Mov Disord, 2020, 35(7): 1208-1217. DOI:10.1002/mds.28052 |
| [13] |
周洪英, 孙波, 吴洪丽, 等. 昆虫肠道微生物功能及家蚕肠道微生物研究进展[J]. 北方蚕业, 2015, 36(4): 1-4, 33. Zhou HY, Sun B, Wu HL, et al. Research progress on function of insects gut microbiota and the microbial of Bombyx mori[J]. North Sericult, 2015, 36(4): 1-4, 33. DOI:10.3969/j.issn.1673-9922.2015.04.001 |
| [14] |
索鹏辉, 符修昊, 安丽萍, 等. 海南省儋州市旱季致倦库蚊肠道细菌多样性研究[J]. 中国热带医学, 2022, 22(1): 20-25. Suo PH, Fu XH, An LP, et al. The diversity of midgut bacteria of Culex quinquefasciatus in the dry season in Danzhou, Hainan[J]. China Trop Med, 2022, 22(1): 20-25. DOI:10.13604/j.cnki.46-1064/r.2022.01 |
| [15] |
夏晓峰. 小菜蛾中肠微生物多样性及其功能研究[D]. 福州: 福建农林大学, 2014. Xia XF. Organizational diversity and functional characterization of microbiota in the midgut of diamondback moth, Plutella xylostella (L.)[D]. Fuzhou: Fujian Agriculture and Forestry University, 2014. (in Chinese) |
| [16] |
Pasti MB, Belli ML. Cellulolytic activity of Actinomycetes isolated from termites (Termitidae) gut[J]. FEMS Microbiol Lett, 1985, 26(1): 107-112. DOI:10.1111/j.1574-6968.1985.tb01574.x |
| [17] |
Waite DW, Taylor MW. Characterizing the avian gut microbiota: Membership, driving influences, and potential function[J]. Front Microbiol, 2014, 5: 223. DOI:10.3389/fmicb.2014.00223 |
| [18] |
Klement RJ, Pazienza V. Impact of different types of diet on gut microbiota profiles and cancer prevention and treatment[J]. Medicina (Kaunas), 2019, 55(4): 84. DOI:10.3390/medicina55040084 |
| [19] |
Boyle DP, Zembower TR. Epidemiology and management of emerging drug-resistant gram-negative bacteria: Extended-spectrum β-lactamases and beyond[J]. Urol Clin North Am, 2015, 42(4): 493-505. DOI:10.1016/j.ucl.2015.05.005 |
| [20] |
Boissière A, Tchioffo MT, Bachar D, et al. Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection[J]. PLoS Pathog, 2012, 8(5): e1002742. DOI:10.1371/journal.ppat.1002742 |
| [21] |
Werren JH. Biology of wolbachia[J]. Ann Rev Entomol, 1997, 42: 587-609. DOI:10.1146/annurev.ento.42.1.587 |
| [22] |
Jaenike J. Spontaneous emergence of a new Wolbachia phenotype[J]. Evolution, 2007, 61(9): 2244-2252. DOI:10.1111/j.1558-5646.2007.00180.x |
| [23] |
Sunantaraporn S, Thepparat A, Phumee A, et al. Culicoides Latreille (Diptera: Ceratopogonidae) as potential vectors for Leishmania martiniquensis and Trypanosoma sp. in northern Thailand[J]. PLoS Negl Trop Dis, 2021, 15(12): e0010014. DOI:10.1371/journal.pntd.0010014 |
| [24] |
Kohl KD. Diversity and function of the avian gut microbiota[J]. J Comp Physiol B, 2012, 182(5): 591-602. DOI:10.1007/s00360-012-0645-z |