 2023, Vol. 34
2023, Vol. 34扩展功能
文章信息
- 臧传慧, 李黎明, 刘烁, 公茂庆, 王文倩, 王奕婷, 娄紫薇, 类晶晶, 程鹏, 刘宏美
- ZANG Chuan-hui, LI Li-ming, LIU Shuo, GONG Mao-qing, WANG Wen-qian, WANG Yi-ting, LOU Zi-wei, LEI Jing-jing, CHENG Peng, LIU Hong-mei
- 基于mtDNA-COⅠ基因的山东省淡色库蚊种群遗传多样性分析
- Genetic diversity of Culex pipiens pallens populations in Shandong province, China based on the mtDNA-COⅠgene
- 中国媒介生物学及控制杂志, 2023, 34(3): 298-302
- Chin J Vector Biol & Control, 2023, 34(3): 298-302
- 10.11853/j.issn.1003.8280.2023.03.003
-
文章历史
- 收稿日期: 2023-01-03




2 济宁市疾病预防控制中心,山东 济宁 272033
2 Jining City Center for Disease Control and Prevention, Jining, Shandong 272033, China
淡色库蚊(Culex pipiens pallens)是山东省流行性乙型脑炎病毒(Japanese encephalitis virus,JEV)和班氏丝虫(Filaria bancrofti)的重要传播媒介,是西尼罗病毒(West Nile virus,WNV)在中国传播的潜在风险媒介,严重威胁着人类的生命健康[1-2],淡色库蚊也是住区常见的骚扰性吸血害虫。目前淡色库蚊最主要的防治手段是使用化学杀虫剂,长期大量的使用杀虫剂使其产生了不同程度的抗药性[3]。研究表明不同地理环境的蚊虫在生态习性、媒介能力和对杀虫剂抗药性等方面均存在一定的差异[4-6]。淡色库蚊不同地理种群遗传结构和遗传分化的研究将有助于解释这些差异。近年来,分子遗传学技术常被应用于研究种群遗传结构,常用的分子标记为微卫星和线粒体基因,其中线粒体细胞色素C氧化酶亚基Ⅰ(mitochondrial cytochrome c oxidase subunitⅠ,mtDNA-COⅠ)基因序列因位置比较保守,母系遗传且遗传进化速率较快,比核DNA更易受遗传漂变的影响,线粒体DNA多态性分析能够在一定层面上了解种群的遗传多样性,还可以探讨形成特定遗传结构的原因,使其成为研究分子进化遗传常用分子标记物,目前已被广泛应用于昆虫的系统进化和种内遗传分化的分析[7-8]。郭秀霞等[9]利用mtDNA-COⅠ基因分析了山东省不同蚊种的遗传多样性。郑梓豪等[10]基于mtDNA-COⅠ基因分析了广州市15个白纹伊蚊(Aedes albopictus)种群的遗传多样性。Chang等[11]基于mtDNA-COⅠ基因序列分析我国中华按蚊(Anopheles sinensis)种群的遗传结构。本文在上述文献报道的基础上,通过采集大量的样本来分析山东省不同淡色库蚊种群的mtDNA-COⅠ基因片段的遗传结构和遗传多样性,为蚊虫系统进化和遗传分化理论的提升提供科学参考,同时为蚊虫治理及抗药性研究提供有价值的见解。
1 材料与方法 1.1 淡色库蚊的采集2020年9月-2022年9月期间,利用灯诱法采集山东省菏泽、德州、聊城、青岛、烟台和日照6个市的淡色库蚊自然种群的现场样本(图 1),采集样本信息见表 1。形态学鉴定方法参考陆宝麟和吴永厚编著的《中国重要医学昆虫分类与鉴别》。经鉴定为淡色库蚊的成蚊,用95%乙醇溶液保存,并置-20 ℃备用。

|
| 图 1 山东省淡色库蚊种群采样地点分布 Figure 1 Distribution of sampling sites of Culex pipiens pallens populations in Shandong |
| |
|
采用DNA提取试剂盒(cadorⓇ Pathogen 96 QIAcubeⓇ HT Kit)按照使用说明提取单只雌性成蚊的基因组DNA。在-80 ℃下保存。
1.3 淡色库蚊mtDNA-COⅠ基因片段的扩增和测序参考Folmer等[12]使用的通用引物序列LCO1490(5'-GGTCAACAAATCATAAAGATATTGG-3')和HCO2198(5'-TAAACTTCAGGGTGACCAAAAAAT CA-3')。PCR反应扩增体系为50 μl,其中2×Phanta Max Master Mix 25 μl,模板DNA 2 μl,正、反向引物(10 μmol/L)各1 μl,ddH2O 21 μl。反应条件:95 ℃ 3 min;95 ℃ 15 s,53 ℃ 15 s,72 ℃ 1 min,30个循环;72 ℃ 7 min,4 ℃保存。出现目的条带的PCR产物送生工生物工程(上海)股份有限公司进行测序。
1.4 数据分析将所获mtDNA-COⅠ基因序列在美国国立生物技术信息中心(NCBI)上经过BLAST比对,与淡色库蚊mtDNA-COⅠ基因匹配度在99%以上的确认为淡色库蚊(分子生物学鉴定)。采用BioEdit 7.0软件对测序结果进行比对处理。采用DnaSP v6[13]软件确定变异位点,计算淡色库蚊各种群的核苷酸多样性(nucleotide diversity,Pi)、单倍型多样性(haplotype diversity,Hd)、中性检验[14]和错配分布分析。应用Arlequin 3.1软件[15]计算各种群间的遗传分化系数(fixation index,FST)和基因流(number of migrants,Nm)及进行分子方差分析(analysis of molecular variance,AMOVA),以明确种群内和种群间的遗传多样性和分化程度。使用PopART 1.7软件绘制单倍型网络图,采用适用于分析种内数据的TCS network方法[16]。
2 结果 2.1 淡色库蚊mtDNA-COⅠ基因片段的碱基组成山东省6个种群共扩增出423条核苷酸序列,每条长度为603 bp。mtDNA-COⅠ序列片段中A、C、G、T平均碱基组成的统计结果显示:(A+T)含量(69.2%) > (G+C)含量(30.8%),表现出AT偏倚特征,符合线粒体DNA的特性。
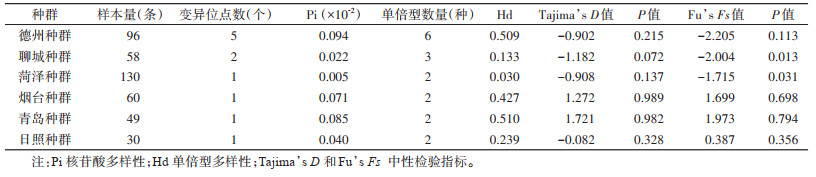
2.2 淡色库蚊mtDNA-COⅠ基因序列特征位点多态性结果显示:603个位点中保守位点有596个,变异位点7个,简约信息位点7个。本研究样本序列共获得8种单倍型,其中不同种群共享单倍型2种,种群特有单倍型6种,H01和H02为共享单倍型,H03~H08分别为各种群的特有单倍型(图 2),序列号分别是OQ600719、OQ603406、OQ603419、OQ603457、OQ603467、OQ603490、OQ603492、OQ603495。整体Pi是0.000 57,Hd是0.333,平均核苷酸差异数(average number of nucleotide difference,k)为0.344。淡色库蚊不同种群mtDNA-COⅠ序列遗传多样性见表 2。

|
| 图 2 基于Median Joining Network构建的淡色库蚊不同种群mtDNA-COⅠ序列单倍型网络图 Figure 2 Haplotype network of mtDNA-COⅠ sequences of different Culex pipiens pallens populations constructed based on the median joining network method |
| |
|
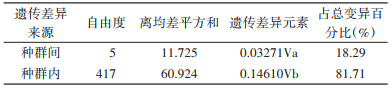
对6个地理种群的淡色库蚊的mtDNA-COⅠ序列进行分子变异等级分析,结果显示:种群内差异为81.71%,种群间差异为18.29%(表 3),种群内变异明显大于种群间变异,差异有统计学意义(P < 0.001),显示遗传分化主要来自于种群内部。
|
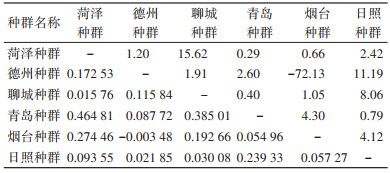
对6个淡色库蚊种群mtDNA-COⅠ序列的FST和Nm值进行计算,种群间FST值为-0.003 48~0.464 81。德州与烟台种群之间的FST值为负数,青岛与菏泽种群之间的FST值最大,为0.464 81,其次是青岛和聊城种群,为0.385 01,与基因流结果相符合。见表 4。
|
中性检验结果(表 2)显示,青岛和烟台种群的Tajima’s D值均为正值,而菏泽、聊城、德州和日照种群的Tajima’s D值均为负值,所有种群Tajima’s D检验均无统计学意义(均P > 0.05)。3个沿海城市青岛、日照和烟台种群的Fu’s Fs值都为正值,而3个内陆城市菏泽、聊城和德州种群的Fu’s Fs值均为负值,只有聊城和菏泽种群的Fu’s Fs检验有统计学意义(P < 0.05)。
3 讨论山东省位于北纬34°~38°之间,而淡色库蚊广泛分布于我国北纬32°以北的多个省份,是我国北方城市的优势蚊种[17]。山东省济南、青岛、淄博、烟台、济宁和泰安市等地调查发现淡色库蚊携带JEV,蚊虫标本感染率在7.58%~17.50%[18]。其种群遗传结构的研究,将对评价其媒介能力和抗药性水平,制定更有针对性的相关传染病防控方案提供重要的指导意义。本次实验采集山东省6个淡色库蚊种群,淡色库蚊Hd为0.030~0.510,Pi为0.000 05~0.000 94。菏泽Hd和Pi最低,德州的Pi最高,青岛的Hd最高。在单倍型网络图中发现,单倍型H01作为中心向其他单倍型发散,推断H01是古老单倍型(由其发出的连线最多,与其他单倍型的联系最为密切)。H01和H02为主要单倍体型,其他单倍体为次要单倍体型。H01为所有种群的共享单倍型,H02为除菏泽外其他5个种群的共享单倍型,H03~H06为德州种群的特有单倍型,H07为菏泽种群的特有单倍型,H08为聊城种群的特有单倍型。
FST表示种群之间的遗传分化程度,范围在0~1之间,值越大表明种群的分化程度越高。一般认为FST为0~0.05,群体间遗传分化很小,可以不考虑其分化;FST为0.05~0.15,群体间存在中等程度的遗传分化;FST为0.15~0.25,群体间遗传分化较大;FST > 0.25,群体间有很大的遗传分化[10]。分子变异分析表明山东省各个市淡色库蚊种群遗传差异很小,遗传差异主要是来自于个体之间。两两比较淡色库蚊的FST表明德州和烟台种群之间无分化;德州和日照、菏泽和聊城、聊城和日照种群之间的遗传分化很小;青岛和菏泽种群间的遗传分化最大,其他种群之间存在中等或者较大的遗传分化。总体来看,青岛种群和大多数种群之间的遗传分化较大,这可能与青岛市采样点位于黄岛有关:多年前黄岛是一个孤岛,随着经济的发展,交通的便利,黄岛和内陆的交流才日渐频繁。
当2种中性检验的统计值接近0时,说明种群处于稳定阶段,而统计值为负值并且有统计学意义时,认为种群在历史上发生过扩张,Tajima’s D检验更倾向于揭示古老种群发生扩张的历史,而Fu’s Fs检验对近期种群的扩张更为敏感。当Tajima’s D值为负且有统计学意义时,可认为不符合中性理论,否则认为符合中性理论,当Fu’s Fs值为负值且有统计学意义时,认为种群在近期经历过扩张,否则认为种群的发展平稳[14]。青岛和烟台种群的Tajima’s D值都为正,菏泽、日照、聊城和德州种群的Tajima’s D值都为负,但所有种群Tajima’s D检验均无统计学意义(P > 0.05),符合中性理论,说明进化过程中选择压力小。青岛、日照和烟台种群的Fu’s Fs检验值都为正值,而菏泽、聊城和德州种群的Fu’s Fs检验值都为负值,其中聊城的Fu’s Fs检验的P值为0.013,菏泽的Fu’s Fs检验的P值为0.031,其他4个种群的Fu’s Fs检验均无统计学意义(P > 0.1),表明聊城和菏泽种群在近期经历过扩张。
蚊虫作为多种疾病的传播媒介,对人类的健康造成了极大的威胁[19-20]。本研究通过对山东省6个淡色库蚊种群的mtDNA-COⅠ基因进行分析,发现了山东省的淡色库蚊部分种群处于群体扩张阶段,两两城市之间的基因交流水平不同,以至于不同城市之间淡色库蚊种群间分化水平也有所差异,有的种群之间不存在遗传分化,有的种群之间存在很大的遗传分化。推断部分种群的媒介能力或抗药性机制可能存在较大差别,因此对媒介蚊虫治理及相关传染病的防控带来很大挑战[21]。
利益冲突 无
| [1] |
Jiang SF, Xing D, Li CX, et al. Replication and transmission of West Nile virus in simulated overwintering adults of Culex pipiens pallens (Diptera: Culicidae) in China[J]. Acta Trop, 2023, 237: 106720. DOI:10.1016/j.actatropica.2022.106720 |
| [2] |
Atoni E, Zhao L, Hu C, et al. A dataset of distribution and diversity of mosquito-associated viruses and their mosquito vectors in China[J]. Sci Data, 2020, 7(1): 342. DOI:10.1038/s41597-020-00687-9 |
| [3] |
Liu HM, Xie LH, Cheng P, et al. Trends in insecticide resistance in Culex pipiens pallens over 20 years in Shandong, China[J]. Parasit Vectors, 2019, 12(1): 167. DOI:10.1186/s13071-019-3416-9 |
| [4] |
吕文祥, 程鹏, 彭荟, 等. 山东省东平湖地区2021年淡色库蚊抗药性调查[J]. 中国媒介生物学及控制杂志, 2022, 33(1): 104-107. Lyu WX, Cheng P, Peng H, et al. Insecticide resistance of Culex pipiens pallens in Dongping Lake area, Shandong province, China, in 2021[J]. Chin J Vector Biol Control, 2022, 33(1): 104-107. DOI:10.11853/j.issn.1003.8280.2022.01.019 |
| [5] |
王海洋, 郭秀霞, 杨秋兰, 等. 鲁西南湿地蚊虫分布及淡色库蚊抗药性调查[J]. 中华卫生杀虫药械, 2020, 26(5): 423-426. Wang HY, Guo XX, Yang QL, et al. Investigation on the distribution of mosquitoes and the insecticide resistance of Culex pipiens pallens in the wetlands of southwest Shandong province[J]. Chin J Hyg Insect Equip, 2020, 26(5): 423-426. DOI:10.19821/j.1671-2781.2020.05.006 |
| [6] |
Liu HM, Yang PP, Cheng P, et al. Resistance level of mosquito species (diptera: Culicidae) from Shandong province, China[J]. Int J Insect Sci, 2015, 7: 47-52. DOI:10.4137/IJIS.S24232 |
| [7] |
Adeniran AA, Hernández-Triana LM, Ortega-Morales AI, et al. Identification of mosquitoes (Diptera: Culicidae) from Mexico state, Mexico using morphology and COⅠ DNA barcoding[J]. Acta Trop, 2021, 213: 105730. DOI:10.1016/j.actatropica.2020.105730 |
| [8] |
Xie GL, Ma XR, Liu QY, et al. Genetic structure of Culex tritaeniorhynchus (Diptera: Culicidae) based on COⅠ DNA barcodes[J]. Mitochondrial DNA B Resour, 2021, 6(4): 1411-1415. DOI:10.1080/23802359.2021.1911711 |
| [9] |
郭秀霞, 程鹏, 刘丽娟, 等. 基于mtDNA-COⅠ基因的山东省蚊种群遗传多样性分析[J]. 中国血吸虫病防治杂志, 2018, 30(1): 37-41. Guo XX, Cheng P, Liu LJ, et al. Analysis of population genetic diversity of mosquitoes from Shandong province based on mitochondrial DNA cytochrome oxidase subunit Ⅰ gene fragment[J]. Chin J Schisto Control, 2018, 30(1): 37-41. DOI:10.16250/j.32.1374.2017091 |
| [10] |
郑梓豪, 吴珊珊, 魏勇, 等. 基于线粒体COⅠ基因分析广州市15个白纹伊蚊种群的遗传多样性[J]. 中国人兽共患病学报, 2021, 37(11): 985-994. Zheng ZH, Wu SS, Wei Y, et al. Analysis of the genetic diversity of 15 Aedes albopictus populations in Guangzhou based on the mitochondrial COⅠ gene[J]. Chin J Zoonoses, 2021, 37(11): 985-994. DOI:10.3969/j.issn.1002-2694.2021.00.141 |
| [11] |
Chang XL, Zhong DB, Lo E, et al. Landscape genetic structure and evolutionary genetics of insecticide resistance gene mutations in Anopheles sinensis[J]. Parasit Vectors, 2016, 9: 228. DOI:10.1186/s13071-016-1513-6 |
| [12] |
Folmer O, Black M, Hoeh W, et al. DNA primers for amplification of mitochondrial cytochrome c oxidase subunitⅠ from diverse metazoan invertebrates[J]. Mol Mar Biol Biotechnol, 1994, 3(5): 294-299. |
| [13] |
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, et al. DnaSP 6:DNA sequence polymorphism analysis of large data sets[J]. Mol Biol Evol, 2017, 34(12): 3299-3302. DOI:10.1093/molbev/msx248 |
| [14] |
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism[J]. Genetics, 1989, 123(3): 585-595. DOI:10.1093/genetics/123.3.585 |
| [15] |
Excoffier L, Lischer HEL. Arlequin suite ver 3.5:A new series of programs to perform population genetics analyses under Linux and windows[J]. Mol Ecol Resour, 2010, 10(3): 564-567. DOI:10.1111/j.1755-0998.2010.02847.x |
| [16] |
Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies[J]. Mol Biol Evol, 1999, 16(1): 37-48. DOI:10.1093/oxfordjournals.molbev.a026036 |
| [17] |
薛志静, 刘小波, 郭玉红, 等. 山东省蚊虫及蚊媒病毒调查研究概况[J]. 中国媒介生物学及控制杂志, 2019, 30(4): 481-484. Xue ZJ, Liu XB, Guo YH, et al. An investigation of mosquitoes and mosquito-borne viruses in Shandong province, China[J]. Chin J Vector Biol Control, 2019, 30(4): 481-484. DOI:10.11853/j.issn.1003.8280.2019.04.032 |
| [18] |
Shi QQ, Song X, Lv YY, et al. Potential risks associated with Japanese encephalitis prevalence in Shandong province, China[J]. Vector Borne Zoonotic Dis, 2019, 19(8): 640-645. DOI:10.1089/vbz.2018.2416 |
| [19] |
Liu HM, Liu LH, Cheng P, et al. Bionomics and insecticide resistance of Aedes albopictus in Shandong, a high latitude and high-risk dengue transmission area in China[J]. Parasit Vectors, 2020, 13(1): 11. DOI:10.1186/s13071-020-3880-2 |
| [20] |
Wu P, Yu X, Wang PH, et al. Arbovirus lifecycle in mosquito: Acquisition, propagation and transmission[J]. Expert Rev Mol Med, 2019, 21: e1. |
| [21] |
Zhang RL, Leng PE, Wang XJ, et al. Molecular analysis and genetic diversity of Aedes albopictus (Diptera: Culicidae) from China[J]. Mitochondrial DNA A DNA Mapp Seq Anal, 2018, 29(4): 594-599. DOI:10.1080/24701394.2017.1325481 |