 2022, Vol. 33
2022, Vol. 33扩展功能
文章信息
- 冯玉亮, 林世华, 潘明, 曹一鸥, 李伟
- FENG Yu-liang, LIN Shi-hua, PAN Ming, CAO Yi-ou, LI Wei
- 四川省首起登革热本地传播疫情登革病毒全基因组特征及溯源分析
- Complete genome characterization and source tracking of Dengue virus from the first local dengue outbreak in Sichuan province, China
- 中国媒介生物学及控制杂志, 2022, 33(2): 239-244
- Chin J Vector Biol & Control, 2022, 33(2): 239-244
- 10.11853/j.issn.1003.8280.2022.02.014
-
文章历史
- 收稿日期: 2021-10-20




登革病毒(Dengue virus)属于黄病毒科,为单股正链RNA病毒。根据血清学特征可分为1、2、3、4四个血清型。登革热(dengue fever)则是由登革病毒引起的一种急性蚊媒传播疾病,传播媒介主要为埃及伊蚊(Aedes aegypti)和白纹伊蚊(Ae. albopictus)[1]。该病广泛流行于热带和亚热带地区,波及全球100多个国家和地区,且发病数仍在不断增加,已成为一个全球性的严重公共卫生问题[2]。我国登革热是输入引起的本地传播或暴发流行,本地传播疫情主要分布于广东、浙江、福建、台湾以及云南省等地[3-5]。四川省常年有零星输入病例,但随着2019年全球登革热疫情暴发[6],输入病例增多,泸州市出现省内首起本地传播暴发疫情。2019年四川省共报告输入登革热病例331例,输入疫源地主要为东南亚国家和国内部分省、市,其中东南亚国家主要为柬埔寨(199例)、泰国(28例)、老挝(10例)、菲律宾(8例)和越南(8例)等;国内输入疫源地主要为广东(8例)和云南省(12例)及重庆市(10例)。泸州市的输入病例疫源地主要为柬埔寨(12例),我国的广东(1例)和云南省(3例)及重庆市(1例)。为了解本次疫情中登革病毒株可能的来源及其分子生物学特征,本研究选取本次疫情中核酸检测阳性的本地病例血清标本扩增登革病毒全基因组序列测序,并进行同源性、系统进化分析和基因组变异分析。
1 材料与方法 1.1 标本来源与检测2019年9月四川省泸州市集中报告了18例疑似登革热病例,分布于泸州市龙马潭和江阳区,且均没有登革热高发地区旅居史。病例标本经ELISA IgM抗体检测和实时荧光定量PCR(RT-qPCR)检测,结果11例为IgM抗体阳性,3例为核酸阳性,基本确认这是1起本地登革热暴发疫情。为了解本次疫情毒株的可能来源及其分子生物学特征,将核酸阳性病例标本重新提取核酸进行全基因组扩增与序列测定。
1.2全基因组扩增和测序
待检病例血清采用病毒总核酸提取试剂盒(Maxwell® 16 Viral Total Nucleic Acid Purification Kit,Promega公司,美国)提取RNA。RNA经反转录试剂盒(Reverse Transcription System,Promega公司,美国)反转录后采用分段引物扩增全基因组,分段引物以GenBank中序列号为KY057373的登革病毒全基因组为模板并参考文献[7]共设计11对引物(表 1)。扩增产物经毛细管电泳鉴定并送成都擎科梓熙生物技术有限公司进行测序。

|
测序完成的11条片段使用DNAStar 5.0软件包中的SeqMan进行拼接和校正;使用BioEdit进行序列比对;使用MegaAlign进行核苷酸和氨基酸的同源性分析;使用MEGA 7.0软件进行进化树分析,选用neighbour-joining(NJ)法,自举重复次数(BP)选择1 000;所有变异分析均以Hawaii株作为参照株[8]。
2 结果 2.1 全基因组测序结果对本次3份病例标本登革病毒全基因组测序完成的片段进行拼接、组装和校正,结果1份标本获得基因组全长序列,2份标本获得包括E基因的全基因组部分序列。经过美国国立生物技术信息中心(NCBI)BLAST分析3份标本中的登革病毒株序列高度一致。将获得基因组全长序列的该株病毒命名为SCD19-218(上传GenBank编号:LC646544),另外获得部分序列的毒株分别命名为SCD19-219和SCD19-220。
2.2 全基因组序列的基本特征本次获得的SCD19-218病毒株全基因组序列全长10 706 bp,4种核苷酸组成分别是:A为33.46%,G为26.30%,T为20.03%,C为20.21%。SCD19-218包含1个连续的开放阅读框(ORF)和两段非编码区。ORF全长10 179 bp(nt 79~10 257)编码3 393个氨基酸,由3个主要结构蛋白和7个非结构蛋白组成。非编码区为ORF左侧78 bp(nt 1~78)和右侧450 bp(nt 10 258~10 706)两段序列。
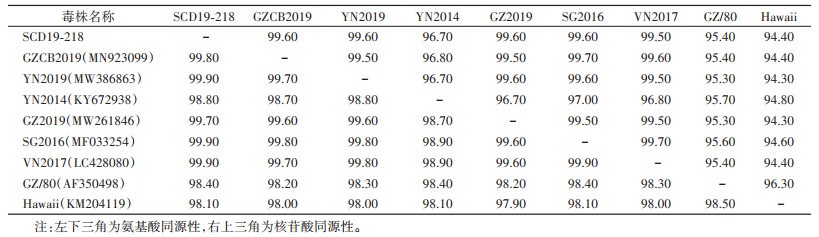
2.3 全基因组同源性分析将SCD19-218与GenBank中收录的部分登革病毒株进行同源性分析(表 2)。结果显示,SCD19-218与广州市分离株GZ2019、广州市分离的柬埔寨输入株GZCB2019、云南省分离株YN2019、新加坡分离株SG2016以及越南分离株VN2017具有较高的同源性,核苷酸和氨基酸同源性分别为99.50%~99.60%和99.70%~99.90%。SCD19-218与登革1型病毒原型株Hawaii的同源性最低,核苷酸和氨基酸同源性分别为94.40%和98.10%。SCD19-218与广州株GZ/80和云南株YN2014的同源性也较低,核苷酸和氨基酸同源性分别为95.40%~96.70%和98.40%~98.80%。
|
选取GenBank数据库中收录的部分登革1~4型病毒株E基因序列和全基因组序列作为参考序列与本次检测的3株毒株序列做进化分析(图 1、2)。E基因系统进化树显示,3株病毒株均属于登革1型GⅠ基因亚型,且彼此之间有相同的进化来源,同属于一个进化分支。在1型毒株GⅠ基因亚型中,SCD19-218、SCD19-219和SCD19-220与2017年越南分离株(LC428079、LC428073)、2016年新加坡分离株(MF033254)、2019年我国广州市分离株(MN945984)、2019年广州市分离的柬埔寨输入株(MN923099)以及2019年云南省分离株(MW386862)的进化距离较近,属于同一进化分支;与原型株Hawaii、泰国分离株(AF425630)以及我国广州市分离株GZ/80株(AF350498)的进化距离最远。全基因组进化分析与E基因进化分析结果相似,SCD19-218与2019年我国广州市分离株(MW261846、MW261839)、2019年云南省分离株(MW386863、MW386864)、广州市分离的柬埔寨输入株(MN923099)、2017年越南分离株(LC428080)以及2016年新加坡分离株(MF033254)进化距离较近;与原型株Hawaii、广州市分离株GZ/80株(AF350498)的进化距离最远;与2014年我国云南省分离株(KY672938)、2019年海南省分离株(MT447147)以及2014年日本分离株(LC011948)的进化距离也相对较远。

|
| 注:▲为四川省本地病例检测毒株。 图 1 登革病毒E基因系统进化树 Figure 1 The phylogenetic tree of the E gene of Dengue virus |
| |

|
| 注:▲为四川省本地病例检测毒株。 图 2 登革病毒全基因组进化树 Figure 2 The phylogenetic tree of the complete genome of Dengue virus |
| |
SCD19-218与登革1型GⅠ基因亚型原型株Hawaii(KM204119)相比,分布着589个核苷酸变异,其中有1个核苷酸缺失并且无插入突变。5'端非编码区有1个核苷酸突变和1个核苷酸缺失,3'端非编码区有13个氨基酸突变。在ORF区域有574个核苷酸突变和64个氨基酸突变,其中结构蛋白区ac蛋白有4个氨基酸突变,PrM蛋白有5个氨基酸突变,E蛋白有12个氨基酸突变;非结构蛋白区NS1蛋白有7个氨基酸突变,NS2蛋白有6个氨基酸突变,NS3蛋白有10个氨基酸突变,NS4蛋白有5个氨基酸突变,NS5蛋白有15个氨基酸突变。
选取与SCD19-218在同一进化分支上的2019年我国云南省分离株(MW386863)、2019年广州市分离株(MW261846)、2016年新加坡分离株(MF033254)以及登革热原型株Hawaii(KM204119)做毒力相关位点区域比对分析(图 3)。结果显示,与原型株相比各流行株均有不同程度的氨基酸位点变异,但在E44、E88、E156、E196、E297、E339、E365、E366、E405、NS3(435)等研究较为清楚的毒力关键位点[9-12]上没有改变;SCD19-218与标准株相比在E52、E227、E272、E312、E324、E380位点发生氨基酸突变;SCD19-218与2019年广州市分离株(MW261846)和2016年新加坡分离株(MF033254)相比在毒力相关位点区域氨基酸序列完全一致;SCD19-218与2019年云南省分离株(MW386863)相比在E蛋白毒力Ⅲ区段E386位氨基酸不一致。

|
| 图 3 SCD19-218毒力相关区域变异位点分析 Figure 3 Variation analysis of virulence-related regions in SCD19-218 |
| |
四川省登革热病例常年以国内高发流行区和东南亚国家输入为主。2019年全球登革疫情暴发,尤其是东南亚国家病例数显著增多,我国多个省份也出现不同程度的流行和暴发[6]。随着输入病例大量增加,四川省出现近年来首起本地传播登革热暴发疫情。为了分析本次疫情病毒株的可能来源,对3份核酸检测阳性病例标本进行登革病毒全基因组扩增并测序,但仅SCD19-218获得全基因组序列,其余2份标本获得部分基因组序列。经基因序列比对,3份病例标本中登革病毒基因序列高度一致,同时E基因进化分析显示3株病毒株有相同的进化来源,推测本次本地疫情的病例可能为同一病毒株的子代病例。分别以3株病毒株E基因和CD19-218全基因组做进化树,结果显示2种分析在进化亚型和与其进化距离近的毒株方面无明显变化,证明选取SCD19-218全基因组为代表分析本轮疫情病毒株的进化关系和基因特征是合理的。
本次本地疫情暴发于泸州市江阳和龙马潭区,流行病学资料显示该区域在疫情暴发前曾有2例来源于柬埔寨的输入病例返乡。另有相关文献报道,病例居住区域蚊媒应急监测布雷图指数为87.93,帐诱指数为2只/(顶·h),为登革热暴发高风险级别[13]。以上提示该区域具备输入疫情本地化传播的条件。对本次检测株序列进行同源性和进化分析,发现SCD19-218、SCD19-219和SCD19-220与2016年新加坡分离株、2017年越南分离株、2019年我国广州市分离株、2019年广州市分离的柬埔寨输入株以及2019年云南省分离株的进化距离最近,同时SCD19-218也与上述毒株有较高的同源性,提示引发本次疫情的毒株可能来源于上述某个地区。结合疫情暴发区输入病例流行病学信息推测,本次疫情可能直接由柬埔寨输入病例引发,其病毒株在柬埔寨、越南、新加坡等东南亚国家长期流行,并在2019年引起国内包括四川省、广州市、云南省等地疫情暴发。SCD19-218与云南省、广州市多年前的分离株(KY672938和GZ/80/AF350498)具有较低的同源性同时进化分支也较远,而广州市分离株GZ/80株被认为是我国本土流行株[14],因此表明SCD19-218来源于国外的流行株而非国内本土流行株,证明了前面输入病毒株本地化传播的观点。2019年四川省输入病例增多,输入疫源地主要为东南亚国家,尤其以柬埔寨为疫源地的输入病例最多(占总病例数一半以上),同时国内其他省、市也有部分病例输入四川省。因此,需要对这些重点地区的流行株做好长期监测,同时加强输入病例的管理。
登革病毒属于RNA病毒,基因组容易变异,目前尚无成熟有效的疫苗上市,因此对登革病毒的变异进行监测对于疫情防控有重要意义。登革病毒变异可分布于各个基因区段,对毒力有影响的区域常分布于编码区,其中研究最多的为E蛋白区。有研究发现,E蛋白有3个毒力区段,分别为:Ⅱ区(包含E52~136和E190~284残基),该区域改变会影响病毒进入细胞和组织偏好性;Ⅲ区(包含E303~395残基),该区段改变会影响病毒膜融合活性;以及Ⅱ区与Ⅲ区段分界面区域(包含E294~305残基)[9]。此外,还有4个毒力相关位点,分别为E44、E156、E405和NS3(435),这些位点改变可对毒力产生影响[10-12]。比对SCD19-218在这些区段内的氨基酸序列,发现其与2019年我国广州市分离株和2016年新加坡分离株完全一致,与2019年云南省分离株也仅有1个非关键位点氨基酸(E386)突变,表明这4株病毒株有相近的来源并在毒力上可能也是一致的。SCD19-218与原型株比较其氨基酸有较多不一致,但在毒力关键氨基酸位点上没有改变,这表明SCD19-218的毒力没有明显变化,只是经过时间演化积累了较多的散在突变。登革病毒非编码区发生突变也可对病毒复制产生影响[15-16],本次四川省流行株SCD19-218在5'非编码区有1个核苷酸突变和1个核苷酸缺失,3'端非编码区有13个氨基酸突变,这些突变是否会引起毒株的增殖和毒力的改变,还有待进一步研究。
多年来四川省登革热疫情均为输入病例,此次本地病例发生说明四川省存在暴发本地疫情的自然条件。因此,加强重点疫源地流行株的跟踪监测、加强输入病例的监测与管理以及加强蚊媒监测等均应作为今后防控的重点工作。
利益冲突 无
| [1] |
张拥军, 吴生根, 王金章, 等. 福建省2015年本地登革热病例的病原学特征[J]. 中国人兽共患病学报, 2019, 35(1): 28-33. Zhang YJ, Wu SG, Wang JZ, et al. Etiological characterization of indigenous dengue cases in Fujian province, 2015[J]. Chin J Zoonoses, 2019, 35(1): 28-33. DOI:10.3969/j.issn.1002-2694.2018.00.210 |
| [2] |
刘起勇. 新时代媒介生物传染病形势及防控对策[J]. 中国媒介生物学及控制杂志, 2019, 30(1): 1-6, 11. Liu QY. Epidemic profile of vector-borne diseases and vector control strategies in the new era[J]. Chin J Vector Biol Control, 2019, 30(1): 1-6, 11. DOI:10.11853/j.issn.1003.8280.2019.01.001 |
| [3] |
刘永华, 尹小雄, 张海林, 等. 云南省德宏州2013-2019年登革热流行特征及媒介伊蚊监测分析[J]. 中国媒介生物学及控制杂志, 2021, 32(2): 173-180. Liu YH, Yin XX, Zhang HL, et al. Epidemiological characteristics of dengue fever and monitoring of Aedes vector mosquitoes in Dehong Dai and Jingpo Autonomous Prefecture of Yunnan province, China, 2013-2019[J]. Chin J Vector Biol Control, 2021, 32(2): 173-180. DOI:10.11853/j.issn.1003.8280.2021.02.011 |
| [4] |
刘媛媛, 刘远, 罗雷, 等. 2011-2019年广州市登革热流行病学特征分析[J]. 现代预防医学, 2021, 48(11): 1925-1929. Liu YY, Liu Y, Luo L, et al. Epidemiological analysis on dengue fever cases in Guangzhou, 2011-2019[J]. Mod Prev Med, 2021, 48(11): 1925-1929. |
| [5] |
Wu WL, Bai ZJ, Zhou HQ, et al. Molecular epidemiology of dengue viruses in southern China from 1978 to 2006[J]. Virol J, 2011, 8(1): 322. DOI:10.1186/1743-422X-8-322 |
| [6] |
李杨, 张文宏. 全球登革热疫情态势、疫情警报[J]. 中华传染病杂志, 2019, 37(10): 619-621. Li Y, Zhang WH. Global dengue epidemic situation and alert[J]. Chin J Infect Dis, 2019, 37(10): 619-621. DOI:10.3760/cma.j.issn.1000-6680.2019.10.008 |
| [7] |
马珍元, 黄呈辉, 张新枝, 等. 2014年深圳市流行的1型登革病毒分子溯源分析[J]. 中国现代医学杂志, 2016, 26(3): 39-44. Ma ZY, Huang CH, Zhang XZ, et al. Molecular phylogenetic analysis of type 1 strains of dengue virus isolated in Shenzhen city during the 2014 dengue outbreak[J]. China J Mod Med, 2016, 26(3): 39-44. DOI:10.3969/j.issn.1005-8982.2016.03.008 |
| [8] |
Tamura K, Stecher G, Peterson D, et al. MEGA 6.0: Molecular evolutionary genetics analysis version 6.0[J]. Mol Biol Evol, 2013, 30(12): 2725-2729. DOI:10.1093/molbev/mst197 |
| [9] |
Guirakhoo F, Zhang Z, Myers G, et al. A single amino acid substitution in the envelope protein of chimeric yellow fever-dengue 1 vaccine virus reduces neurovirulence for suckling mice and viremia/viscerotropism for monkeys[J]. J Virol, 2004, 78(18): 9998-10008. DOI:10.1128/JVI.78.18.9998-10008.2004 |
| [10] |
赵卫, 杨佩英. 登革病毒毒力研究进展[J]. 中华实验和临床病毒学杂志, 2001, 15(2): 197-199. Zhao W, Yang PY. Research progress on virulence of dengue virus[J]. Chin J Exp Clin Virol, 2001, 15(2): 197-199. DOI:10.3760/cma.j.issn.1003-9279.2001.02.037 |
| [11] |
Holmes EC, Worobey M, Rambaut A. Phylogenetic evidence for recombination in dengue virus[J]. Mol Biol Evol, 1999, 16(3): 405-409. DOI:10.1093/oxfordjournals.molbev.a026121 |
| [12] |
Dos Santos CND, Frenkiel MP, Courageot MP, et al. Determinants in the envelope E protein and viral RNA helicase NS3 that influence the induction of apoptosis in response to infection with dengue type 1 virus[J]. Virology, 2000, 274(2): 292-308. DOI:10.1006/viro.2000.0457 |
| [13] |
曹一鸥, 吕强, 张佳珂, 等. 2019年四川省首例登革热本地病例调查与疫区处置情况分析[J]. 预防医学情报杂志, 2020, 36(11): 1499-1503. Cao YO, Lyu Q, Zhang JK, et al. Investigation and disposal of the first local case of dengue fever in Sichuan province[J]. J Prev Med Inf, 2020, 36(11): 1499-1503. |
| [14] |
李晓萸, 耿丽卿, 苑锡同, 等. 我国登革1型病毒广州株基因组全序列测定与分析[J]. 军事医学科学院院刊, 2001, 25(3): 161-165. Li XY, Geng LQ, Yuan XT, et al. Determination and analysis of the complete genome of dengue 1 virus GZ/80 strain[J]. Bull Acad Mil Med Sci, 2001, 25(3): 161-165. DOI:10.3969/j.issn.1674-9960.2001.03.001 |
| [15] |
Cahour A, Pletnev A, Vazeille-Falcoz M, et al. Growth-restricted dengue virus mutants containing deletions in the 5'noncoding region of the RNA genome[J]. Virology, 1995, 207(1): 68-76. DOI:10.1006/viro.1995.1052 |
| [16] |
Men R, Bray M, Clark D, et al. Dengue type 4 virus mutants containing deletions in the 3'noncoding region of the RNA genome: Analysis of growth restriction in cell culture and altered viremia pattern and immunogenicity in rhesus monkeys[J]. J Virol, 1996, 70(6): 3930-3937. DOI:10.1128/JVI.70.6.3930-3937.1996 |