 2022, Vol. 33
2022, Vol. 33扩展功能
文章信息
- 刘文娟, 张可心, 程鹏, 张心雨, 张倩, 张忠, 张瑞玲
- LIU Wen-juan, ZHANG Ke-xin, CHENG Peng, ZHANG Xin-yu, ZHANG Qian, ZHANG Zhong, ZHANG Rui-ling
- 白纹伊蚊性别差异miRNA筛选及miRNA-mRNA调控网络分析
- Deciphering sex differentially expressed miRNAs and miRNA-mRNA regulatory networks of Aedes albopictus
- 中国媒介生物学及控制杂志, 2022, 33(2): 191-200
- Chin J Vector Biol & Control, 2022, 33(2): 191-200
- 10.11853/j.issn.1003.8280.2022.02.005
-
文章历史
- 收稿日期: 2021-10-02




2 山东第一医科大学寄生虫病防治研究所, 山东 济宁 272033
2 Shandong Institute of Parasitic Diseases, Shandong First Medical University, Jining, Shandong 272033, China
全球超过80%的人口面临着媒介生物传染病的风险,其中蚊虫传播的疾病是给人类健康造成严重负担的主要媒介生物传染病[1]。白纹伊蚊(Aedes albopictus),又称亚洲虎蚊,隶属于双翅目(Diptera)、蚊科(Culicidae)、伊蚊属,是世界上重要的虫媒病毒传播媒介之一。该蚊虫起源于东南亚,其卵具有很强的抗寒、抗旱能力和生命力,容易被携带传播,现已扩散至全球70多个国家和地区,在我国分布也十分广泛[2]。作为登革病毒、基孔肯雅病毒、黄热病病毒等多种虫媒病毒的传播媒介,白纹伊蚊在世界范围内的扩散给全球公共卫生造成了严重威胁[3]。
目前广为研究的小分子RNA(small RNA,sRNA)主要包括microRNA(miRNA)、short interfering RNA(siRNA)和PIWI-interacting RNA(piRNA)。miRNA是一类长约23 nt的内源性非编码小分子RNA,是一种重要的转录后调节剂。在动物体内,miRNA基因经RNA聚合酶Ⅱ作用形成miRNA初级转录产物(primary miRNA,pri-miRNA),pri-miRNA经进一步切割生成RNA前体,Dicer酶切割RNA前体形成成熟的miRNA复合体,Argonaute-1(Ago-1)蛋白与其中一条链形成RNA介导的沉默复合体(RNA induced silencing complex,RISC),另一条链通常会迅速降解消失[4]。在RISC的引导下,miRNA可与靶基因3'非编码区(3'untranslated region,3'UTR)碱基互补配对,或与5' UTR和编码区(coding sequence,CDS)相结合,促进翻译抑制和RNA特异性降解,从而发挥其调控功能[5]。在动物中,miRNA采用“种子区”(seed region)识别靶基因,从而对靶基因进行调控。“种子区”是miRNA 5'端2~8位的一段核苷酸序列。由于miRNA与靶基因的结合并非完全互补配对,因此1个miRNA可能调控多个mRNA,1个mRNA也可能受不同miRNA的共同调控[4]。
蚊虫传播病毒主要有垂直传播和水平传播2种方式,垂直传播是雌蚊通过产卵将病毒传给子代;水平传播主要通过雌蚊叮咬易感人群传播。此外,经垂直传播感染病毒的雄蚊与未感染的雌蚊交配后也可将病毒传递给雌蚊及其子代[6]。雌蚊可通过垂直传播(产卵)和水平传播(叮咬宿主吸血)2种方式传播病原体,在病毒的传播过程中起到了关键作用。本研究旨在通过雌、雄白纹伊蚊差异miRNA和miRNA-mRNA互作网络关系和功能富集分析,探讨白纹伊蚊雌、雄样品间差异表达的miRNA以及其所调控的mRNA,进而筛选鉴定可能参与白纹伊蚊生殖发育调控的候选基因,为进一步明确候选基因的功能奠定基础。
1 材料与方法 1.1 样本收集白纹伊蚊为2017年采自山东省淄博市,在实验室内持续饲养的敏感株。实验室内置于温度为(26±1)℃、湿度为(75±5)%、光周期14 h:10 h的环境条件下饲养。幼蚊饲以猪肝粉,成蚊饲以10%的葡萄糖液,并用小白鼠(昆明小鼠,KM Mouse;雄性,约40 g,普通级动物房饲养)提供血餐,以保证雌蚊可以产卵传代。雌、雄白纹伊蚊从蛹期开始分开饲养,收集羽化后2~3 d未进食的雌、雄成蚊各10只作为本研究的样本。
1.2 miRNA文库的构建和测序 1.2.1 总RNA提取分别取雌、雄成蚊,各分为3组(雌:F1、F2、F3;雄:M1、M2、M3),每组3只样品。用TRIZOL(Invitrogen,美国)方法提取样品总RNA,琼脂糖凝胶电泳分析RNA降解程度以及是否有污染,使用Nanodrop(IMPLEN,美国)检测RNA的纯度(A260/A280比值,A为吸光度),用Qubit(Life Technologies,美国)对RNA浓度进行精确定量,用Agilent 2100(Agilent Technologies,美国)精确检测RNA的完整性。
1.2.2 miRNA文库构建通过构建sRNA文库获得雌、雄白纹伊蚊miRNA信息。样品检测合格后,使用NEBNext®MultiplexSmall RNA Library Prep Set for Illumina®(NEB,美国)构建文库,文库质量检验合格后,利用IlluminaSE50测序,得到miRNA序列信息。
1.2.3 mRNA测序及分析采用去除核糖体RNA的方法构建链特异性文库[7]得到雌、雄白纹伊蚊mRNA测序信息。库检合格后,根据文库的有效浓度及数据产出需求pooling后进行Illumina PE150测序,得到mRNA序列信息。
1.3 miRNA和mRNA差异表达分析为了进行雌、雄白纹伊蚊2个组别间miRNA的表达水平分析,对各样本中已知miRNA和新miRNA进行表达量的统计,并用Transcripts per million(TPM)[8]进行表达量归一化处理。公式:
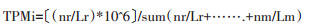
|
nr:比对到目标基因上的序列(read)数;Lr:目标基因外显子长度的总和。
测序得到的原始序列数数据,采用基于负二项分布的DESeq2-1.2.10[9]对白纹伊蚊雌、雄样品miRNA进行差异表达分析,本研究中差异miRNA的筛选条件为:校正后P值(padj)<0.05&|log2(foldchange)|>0.8。
将得到的mRNA进行表达水平定量,表达水平用Fragments per kilobase of transcript sequence per millions base pairs sequenced(FPKM)表示。FPKM是每百万片段中来自某一基因/转录本每千碱基长度的片段数目,该方法同时校正了测序深度和基因长度对片段计数的影响,是目前最为常用的基因表达水平估算方法。本研究中差异mRNA的筛选条件为:padj<0.05&|log2(foldchange)|>1.3。
1.4 miRNAs靶基因预测及功能富集分析采用miRanda-3.3a[10]和RNAhybrid v2.0[11]软件预测miRNA的靶基因,取2种预测结果的交集,进而得到miRNA和靶基因间的对应关系。利用Goseq-Release 2.12软件进行基因本体论(gene ontology,GO)(http://www.geneontology.org/)富集分析,基于Wallenius non-central hyper-geometric distribution,从而更准确地计算出GO term被候选靶基因富集的概率[12]。对基因功能的描述从生物过程(biological process,BP)、细胞组分(cellular component,CC)和分子功能(molecular function,MF)3个方面进行,得到基因的注释信息。采用KOBAS-v2.0软件进行京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)[13]通路显著性富集分析,应用超几何检验找出与整个基因组背景相比在候选靶基因中显著性富集的通路。通过通路显著性富集确定候选靶基因参与的最主要生化代谢途径和信号转导途径。
1.5 miRNA-mRNA关联分析分别筛选白纹伊蚊雌、雄样品间差异表达的miRNA和mRNA,再将雌、雄样品间差异表达的miRNA所对应的靶基因(mRNA)与雌、雄样品间差异表达的mRNA取交集,进而得到用于关联分析的mRNA。根据miRNA对mRNA的调控作用原理,筛选显著下调的miRNA和显著上调的mRNA,以及显著上调的miRNA和显著下调的mRNA的组合作为靶基因对。将上述标准筛选得到的miRNA-mRNA数据输入Cytoscape-3.8.2中作图,得到miRNA-mRNA互作关系网络图。
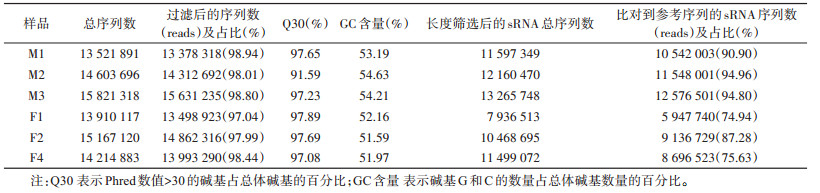
2 结果 2.1 测序结果概述为了获得白纹伊蚊雌、雄样品中miRNA的表达特征,本研究分别构建了雌性样品(F1、F2、F3)和雄性样品(M1、M2、M3)共6个sRNA测序文库。每个文库平均获得14 539 837.5个原始reads,去除带接头的、低质量的reads后,每个文库平均得到14 279 462.3个过滤后的序列数(clean reads)。为定位雌、雄样品中miRNA在基因组上的分布,对各样品的clean reads进行筛选,选取18~40 nt长度的片段miRNA用于后续分析。利用bowtie-0.12.9[14]将长度筛选后的miRNA定位到已发表的白纹伊蚊参考基因组(www.ncbi.nlm.nih.gov/genome/45,Genome version:AalbF2,assembly:GCA_006496715.1)[3]序列上,分析sRNA在参考序列上的分布情况,并注释。将筛选出的clean reads与已公布的白纹伊蚊基因组比对后,发现6个文库中74.94%~94.96%的clean reads与基因组匹配。见表 1。
|
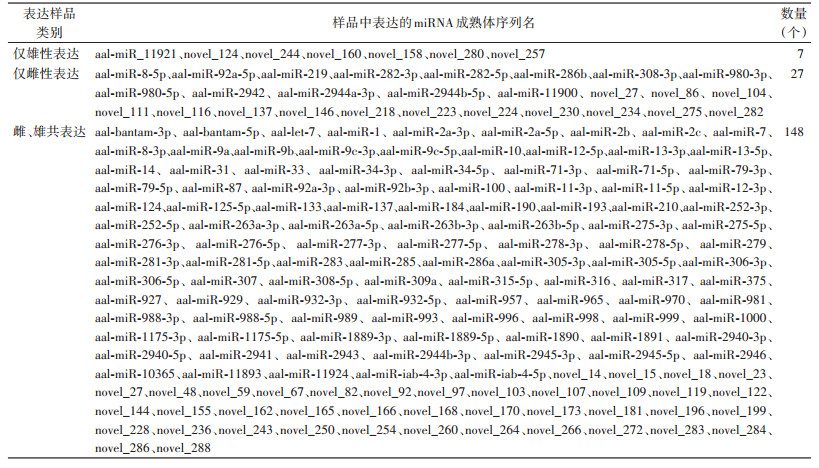
将上述比对到参考基因组上的reads与miRBase数据库中序列进行比对,得到各样品匹配上的sRNA的详细情况,包括匹配上的已知miRNA的二级结构,各样本中miRNA的序列、长度、出现的次数等信息。基于miRNA前体的标志性发夹结构预测新的miRNA。联合使用miREvo_v1.1[15]和mirdeep2_0_0_5[16]预测软件进行新miRNA的分析,通过截取一定长度sRNA比对上的参考序列,探寻其二级结构及Dicer酶切位点信息、能量等特征进行分析,预测样品中新miRNA,并得到各样本中匹配上的sRNA的序列、长度、出现的次数等信息。结果显示,6个miRNA文库共检测到122个已知miRNA成熟体和60个新miRNA成熟体。在这182个检测到的miRNA成熟体中,7个miRNA仅在雄性中表达,27个miRNA仅在雌性中表达;其余的148个miRNA在雄性和雌性白纹伊蚊中都表达。见表 2。
|
差异表达基因分析是比较转录组测序研究的重要环节,也是挖掘重要调控miRNA的基础,从中寻找关键基因进而解释其生物学的分子调控机制。在构建文库的基础上,分析雌、雄两性差异表达的miRNA和mRNA,进一步利用生物信息学方法预测与注释差异表达miRNA的靶基因,分析miRNA和靶基因的表达模式,同时与mRNA测序数据进行联合分析,为探究miRNA调控白纹伊蚊性别分化的分子机制提供更多信息。
2.2.1 差异表达miRNA和差异表达mRNA的分析与鉴定差异分析结果表明,雌、雄样品间共检测到74个差异性表达miRNA,包括39个上调的miRNA(novel_260、aal-miR-999、aal-miR-11-3p、aal-miR-988-5p、aal-miR-932-3p等),其中包含23个已知miRNA和16个新miRNA;35个下调的miRNA(aal-miR-2941、aal-miR-283、aal-miR-998、aal-miR-iab-4-5p、aal-miR-2946等),其中包含29个已知miRNA和6个新miRNA(图 1A)。对74个差异表达miRNA进行聚类分析,结果显示39个上调的miRNA在雄蚊中显著高表达,在雌蚊中显著低表达;35个下调的miRNA在雌蚊中显著高表达,在雄蚊中显著低表达(图 1B)。值得注意的是,74个差异表达miRNA中,5个miRNA仅在雄蚊中检测到表达(novel_160、novel_244、novel_280、novel_257、novel_158);3个miRNA仅在雌蚊中检测到表达(aal-miR-980-3p、novel_104、novel_274)。

|
| 注:红色圆点表示上调表达的miRNA或mRNA;绿色圆点表示下调表达的miRNA或mRNA;蓝色圆点表示无显著差异的miRNA或mRNA;红色和绿色条带分别表示miRNA或mRNA的显著高表达和显著低表达;M代表雄蚊;F代表雌蚊;1、2、3代表不同的样品组数;padj调整后的P值;fold change表示样品表达量的差异倍数;up和up regulated上调;down和down regulated下调。 图 1 miRNA和mRNA差异表达火山图和热图 Figure 1 Volcanic map and heat maps of differentially expressed miRNA and mRNA |
| |
此外,雌、雄白纹伊蚊共检测到1 983个差异表达的mRNA,包括1 021个上调表达的mRNA和962个下调表达的mRNA(图 1C)。对前20个最显著的差异表达mRNA进行聚类分析,结果以热图的形式展示(图 1D)。
2.2.2 差异表达miRNA靶基因预测及GO和KEGG功能富集分析根据miRNA与其靶基因间的对应关系,基于靶基因预测软件,本研究筛选到74个差异表达miRNA对应的16 646个靶基因。为了进一步探究雌、雄白纹伊蚊差异miRNA的功能,对差异表达miRNA的靶基因分别进行GO和KEGG功能富集分析。
差异表达miRNA靶基因的GO功能富集分析中,最具代表性的生物学途径(biological process,BP)条目为氧化还原过程(GO:0055114),其次是跨膜运输(GO:0055085)(图 2A);最具有代表性的细胞学组件(cellular component,CC)条目为核染色质(GO:0000785),其次分别为DNA装配复合体(GO:0044815)、DNA-蛋白质复合体(GO:0032993)、核小体(GO:0000786)(图 2B);分子功能(molecular function,MF)条目中,最具有代表性的为催化活性(GO:0003824),其次分别是离子结合(GO:0043167)、水解酶活性(GO:0016787)、阳离子绑定(GO:0043169)、氧化还原酶活性(GO:0016491)(图 2C)。

|
| 注:GO表示基因本体论数据库(gene ontology, GO);GeneRatio表示差异基因中与该Term相关的基因数与整个差异基因总数的比值;count和Gene_numer均表示富集基因数目;KEGG为京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG);Rich factor指差异表达的基因中位于该代谢路径下差异基因数目与所有注释到该路径基因数目的比值;q value表示经过多重校正的P值。 图 2 雌、雄白纹伊蚊差异表达miRNA靶基因的GO功能富集和KEGG通路富集分析结果 Figure 2 The GO term and KEGG pathway function enrichment results of target genes of miRNAs differentially expressed in female and male samples |
| |
KEGG富集分析的结果显示,最显著富集的通路是糖鞘脂生物合成-神经节系列(aag00604),其次分别为昼夜节律-蝇(aag04711)、调节自噬(aag04140)、其他多糖降解(aag00511)、有机含硒化合物新陈代谢(aag00450)(图 2D)。
2.3 miRNA-mRNA关联分析 2.3.1 雌、雄白纹伊蚊差异表达miRNA-mRNA互作网络分析基于所筛选到的雌、雄两性样品中39个上调的miRNA和35个下调的miRNA,以及相对应的负调控靶基因:388个下调的mRNA和484个上调的mRNA,对二者进行关联分析,得到miRNA-mRNA的负表达调控模式。
为进一步探讨miRNA-mRNA的负调控模式,对雌、雄差异表达miRNA及其对应的负调控靶基因进行分析,得到雌性(图 3A)和雄性(图 3B)高表达miRNA-mRNA互作关系网络图,并基于网络关系中节点的关联度,在2个网络关系图中分别筛选到20个节点关联度最高的核心基因(hub gene)(表 3)。为了进一步探究hub gene之间的表达和调控关系,又分别构建雌性(图 3C)和雄性(图 3D)hub gene互作网络图。

|
| 注:up上调;down下调;图中蓝色标签表示核心基因(hub gene)名称。 图 3 白纹伊蚊高表达miRNA-mRNA和hub gene互作网络图 Figure 3 The interactions network maps of highly expressed miRNA-mRNA and hub gene in Aedes albopictus |
| |

|
雌性高表达miRNA-mRNA互作网络图中,共包含470对雌性特异表达的miRNA-mRNA(miRNA下调,mRNA上调),节点关联度最高的miRNA为aal-miR-998和aal-miR-375,二者均直接参与调控30个mRNA(LOC109406040、XLOC_166385、XLOC_130168、XLOC_026211、XLOC_025154等)(图 3A)。节点关联度最高的mRNA为XLOC_026211,受24个miRNA(aal-miR-283、aal-miR-375、aal-miR-278-3p、aal-miR-193、aal-miR-1175-3p等)直接调控(图 3A)。在雌性hub gene互作网络图中,aal-miR-998和aal-miR-375均参与调控5个重要的mRNA,其中XLOC_026211、XLOC_015703和XLOC_166385受二者共同调控。aal-miR-278-3p、aal-miR-305-5p、aal-miR-980-3p等9个miRNA共同参与调控XLOC_026211(图 3C)。
雄性高表达miRNA-mRNA互作网络图中,共包含481对雄性特异表达的miRNA-mRNA(miRNA上调,mRNA下调),节点关联度最高的mRNA为LOC115258036,受29个miRNA(aal-miR-7、novel_244、aal-miR-71-5p、aal-miR-305-3p、aal-miR-13-5p等)直接调控(图 3B)。节点关联度最高的miRNA是novel_158,直接参与调控27个mRNA(LOC109433010、LOC115270160、LOC109433010、XLOC_067934、XLOC_144664等)(图 3B)。在雄性hub gene互作网络图中,节点关联度最高的mRNA为LOC115258036,受13个重要的mRNA(aal-miR-87、novel_165、novel_48、novel_147、novel_168等)共同调控。Hub gene中节点关联度最高的miRNA是novel_165,同时参与调控5个重要的mRNA(LOC11525803、XLOC_063708、XLOC_021938、XLOC_003885、XLOC_00005)(图 3D)。
2.3.2 雌、雄白纹伊蚊差异表达miRNA负调控靶基因的功能富集分析为了进一步阐明雌、雄白纹伊蚊miRNA-mRNA调控网络的表达模式及功能,分别对2个网络图中miRNA调控的靶基因进行GO功能和KEGG通路富集分析(图 4)。

|
| 注:GO表示基因本体论数据库(gene ontology, GO);GeneRatio表示差异基因中与该Term相关的基因数与整个差异基因总数的比值;Count和Gene_number均表示富集基因数目;KEGG表示京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG);Rich factor指差异表达的基因中位于该代谢路径下差异基因数目与所有注释到该路径基因数目的比值;q value表示经过多重校正的P值。 图 4 白纹伊蚊高表达miRNA负调控靶基因的GO和KEGG功能富集结果 Figure 4 GO terms and KEGG enrichment pathways of target mRNAs that negative regulatory by highly expressed miRNAs in Aedes albopictus |
| |
雌蚊中显著高表达miRNA负调控靶基因的GO富集通路中,最具代表性的条目为膜(GO:0016020),其次分别为定位(GO:0051179)、蛋白质代谢过程(GO:0019538)、运输活性(GO:0005215)、蛋白水解作用(GO:0006508)(图 4A)。值得关注的是,通过GO分析富集到了雌性生殖和性别分化功能相关的条目,分别为:雌性性别分化(GO:0046660)、性别分化(GO:0007548)、精子与透明带的结合(GO:0007339)、精卵识别(GO:0035036)和有性生殖(GO:0019953)。雌蚊显著高表达miRNA负调控靶基因的KEGG富集通路中,最具代表性的通路为核黄素的新陈代谢(aag00740),其次分别为昼夜节律-蝇(aag04711)、精氨酸和脯氨酸代谢(aag00330)、氮代谢(aag00910)和谷胱甘肽代谢(aag00480)(图 4C)。
雄蚊中显著高表达miRNA负调控靶基因的GO功能富集中,最具代表性的条目是水解酶活性(GO:0016787),其次分别为胞内细胞器的部分(GO:0044446)、细胞器的部分(GO:0044422)、蛋白水解作用(GO:0006508)、作用于L-氨基肽的肽酶活性(GO:0070011)(图 4B)。值得关注的是,GO分析中富集到雄性生殖功能相关的条目,分别为无性孢子形成(GO:0030436)、无性生殖(GO:0019954)、多细胞生物的繁殖过程(GO:0048609)、单个生物体的繁殖过程(GO:0044702)、有性生殖(GO:0019953)、多个生物体的繁殖过程(GO:0044703)和生殖过程(GO:0022414)。雄性白纹伊蚊显著高表达miRNA负调控靶基因的KEGG富集通路中,最具代表性的通路为氮代谢(aag00910),其次分别为黏蛋白型o-聚糖的生物合成(aag00512)、酪氨酸代谢(aag00350)、不饱和脂肪酸的生物合成(aag01040)和萜类化合物生物合成支柱(aag00900)(图 4D)。
3 讨论本研究通过雌、雄白纹伊蚊中的miRNA表达谱的分析,进一步结合miRNA调控的mRNA靶基因信息,探讨了参与白纹伊蚊性别决定及生殖过程相关的潜在基因。通过对miRNA进行鉴定和分类,本研究共鉴定出122个已知miRNA成熟体和60个新miRNA成熟体(表 2),发现78个在雌、雄样本间差异表达的miRNAs(图 1A),其中包含8个性别特异性miRNA,这些miRNA可能在雌、雄白纹伊蚊性别决定和发育过程中起关键作用。通过预测差异表达miRNA对应的靶基因,进一步探讨差异表达miRNA的功能。GO富集分析的结果表明,雌、雄白纹伊蚊差异表达miRNA潜在的靶基因主要与氧化还原(GO:0055114)、跨膜运输(GO:0055085)等功能相关(图 2A),提示雌、雄差异表达miRNA可能主要参与能量代谢和发育过程的调节。
通过差异表达miRNA的层次聚类分析发现,39个上调表达的miRNA在雄性白纹伊蚊中显著高表达,在雌性白纹伊蚊中显著低表达;35个下调表达的miRNA在雌性白纹伊蚊中显著高表达,在雄性白纹伊蚊中显著低表达(图 1B)。分别对雌、雄性显著高表达miRNA进行负调控mRNA靶基因的筛选,随后进行miRNA-mRNA互作网络分析,并结合靶基因进行GO功能和KEGG通路富集分析。GO富集的条目表明,雌、雄白纹伊蚊显著高表达miRNA潜在的靶基因与蛋白质分解及代谢功能相关(图 4A、B)。
雌性白纹伊蚊显著高表达miRNA及其负调控mRNA靶基因网络中,hub gene XLOC_166385同时受15个miRNA调控(aal-miR-31、novel_23、aal-miR-9a、novel_199、aal-miR-2945-3p、aal-bantam-3p、novel_18、aal-miR-998、aal-miR-996、aal-miR-305-5p、aal-miR-980-3p、aal-miR-276-3p、aal-miR-9b、aal-miR-9c-5p、aal-miR-375)(图 3A),其中包括3个hub gene:aal-miR-998、aal-miR-375、aal-miR-980-3p(图 3C)。功能富集结果显示,XLOC_166385与雌性性别分化(GO:0046660)、性别分化(GO:0007548)功能相关。提示aal-miR-998、aal-miR-375、aal-miR-980-3p可能参与雌性白纹伊蚊性别分化过程的调控。
功能富集结果显示,LOC109399373和精子与透明带的结合(GO:0007339)、精卵识别(GO:0035036)、有性生殖(GO:0019953)功能相关,该基因受aal-miR-31单独调控(图 3A)。而aal-miR-31同时调控XLOC_166385和LOC109399373基因,提示aal-miR-31可能参与雌性白纹伊蚊性别分化及生殖过程的调控。
雄性白纹伊蚊显著高表达miRNA及其负调控mRNA靶基因网络中,LOC109403788受hub gene aal-miR-34-5p单独调控(图 3B)。此外,aal-miR-34-5p还参与调控4个hub gene(XLOC_063708、XLOC_021938、XLOC_003885、XLOC_000051)(图 3D)。功能富集结果表明,LOC109403788与无性孢子形成(GO:0030436)及无性生殖(GO:0019954)功能有关。推测aal-miR-34-5p可能通过调控LOC109403788参与雄性白纹伊蚊生殖过程的调节,在雌性生殖功能和发育过程中起到重要作用。
功能富集结果表明,LOC109404958与多细胞生物的繁殖过程(GO:0048609)、单个生物体的繁殖过程(GO:0044702)、有性生殖(GO:0019953)、多个生物体的繁殖过程(GO:0044703)和生殖过程(GO:0022414)功能有关。LOC109404958受aal-miR-350-3p单独调控(图 3B),提示aal-miR-350-3p可能通过对LOC109404958的调控,在雄性白纹伊蚊性别决定中起作用。
miRNA-mRNA互作网络图分析丰富了我们对非模式生物白纹伊蚊中miRNA的负调控模式及靶向特异性的认识,为进一步了解白纹伊蚊miRNAs的调控功能提供了相关信息。此外,本研究中检测到了4个雌、雄白纹伊蚊性别分化和生殖功能相关的mRNA,其中XLOC_166385是本研究中新发现的mRNA,而LOC109399373、LOC109403788、LOC109404958早在其他研究中得到发现[17]。通过互作网络分析获得了参与调控这些mRNA的miRNA(图 3A、B),这些miRNA值得我们进一步研究。
利益冲突 无
| [1] |
Franklinos LHV, Jones KE, Redding DW, et al. The effect of global change on mosquito-borne disease[J]. Lancet Infect Dis, 2019, 19(9): e302-e312. DOI:10.1016/S1473-3099(19)30161-6 |
| [2] |
杨舒然, 刘起勇. 白纹伊蚊的全球分布及扩散趋势[J]. 中国媒介生物学及控制杂志, 2013, 24(1): 1-4. Yang SR, Liu QY. Trend in global distribution and spread of Aedes albopictus[J]. Chin J Vector Biol Control, 2013, 24(1): 1-4. |
| [3] |
Palatini U, Masri RA, Cosme LV, et al. Improved reference genome of the arboviral vector Aedes albopictus[J]. Genome Biol, 2020, 21(1): 215. DOI:10.1186/s13059-020-02141-w |
| [4] |
张强. 桔小实蝇发育阶段转变前后miRNA分析及miR-309a调控卵巢发育的分子机制[D]. 重庆: 西南大学, 2020. DOI: 10.27684/d.cnki.gxndx.2020.000080. Zhang Q. miRNA analysis of transitions between different developmental stages and the molecular mechanism of miR-309a regulating the ovarian development in Bactrocera dorsalis[D]. Chongqing: Southwest University, 2020. DOI: 10.27684/d.cnki.gxndx.2020.000080. (inChinese) |
| [5] |
Bushati N, Cohen SM. microRNA functions[J]. Annu Rev Cell Dev Biol, 2007, 23: 175-205. DOI:10.1146/annurev.cellbio.23.090506.123406 |
| [6] |
赵星, 左丽. 伊蚊对登革病毒的垂直与水平传播[J]. 国外医学•流行病学传染病学分册, 2004, 31(2): 108-110. Zhao X, Zuo L. Vertical and horizontal transmission of dengue virus by Aedes[J]. Epidemiol Lemol Foreign Med Sci, 2004, 31(2): 108-110. DOI:10.3760/cma.j.issn.1673-4149.2004.02.015 |
| [7] |
Parkhomchuk D, Borodina T, Amstislavskiy V, et al. Transcriptome analysis by strand-specific sequencing of complementary DNA[J]. Nucleic Acids Res, 2009, 37(18): e123. DOI:10.1093/nar/gkp596 |
| [8] |
Zhou L, Chen JH, Li ZZ, et al. Integrated profiling of microRNAs and mRNAs: MicroRNAs located on Xq27.3 associate with clear cell renal cell carcinoma[J]. PLoS One, 2010, 5(12): e15224. DOI:10.1371/journal.pone.0015224 |
| [9] |
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2[J]. Genome Biol, 2014, 15(12): 550. DOI:10.1186/s13059-014-0550-8 |
| [10] |
Enright AJ, John B, Gaul U, et al. MicroRNA targets in Drosophila[J]. Genome Biol, 2003, 5(1): R1. DOI:10.1186/gb-2003-5-1-r1 |
| [11] |
Krüger J, Rehmsmeier M. RNAhybrid: MicroRNA target prediction easy, fast and flexible[J]. Nucleic Acids Res, 2006, 34(Suppl 2): W451-W454. DOI:10.1093/nar/gkl243 |
| [12] |
Young MD, Wakefield MJ, Smyth GK, et al. Gene ontology analysis for RNA-seq: Accounting for selection bias[J]. Genome Biol, 2010, 11(2): R14. DOI:10.1186/gb-2010-11-2-r14 |
| [13] |
Kanehisa M, Araki M, Goto S, et al. KEGG for linking genomes to life and the environment[J]. Nucleic Acids Res, 2008, 36 Suppl 1: D480-D484. DOI:10.1093/nar/gkm882 |
| [14] |
Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome[J]. Genome Biol, 2009, 10(3): 1-10. DOI:10.1186/gb-2009-10-3-r25 |
| [15] |
Wen M, Shen Y, Shi SH, et al. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments[J]. BMC Bioinformatics, 2012, 13: 140. DOI:10.1186/1471-2105-13-140 |
| [16] |
Friedländer MR, Mackowiak SD, Li N, et al. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades[J]. Nucleic Acids Res, 2012, 40(1): 37-52. DOI:10.1093/nar/gkr688 |
| [17] |
Poelchau MF, Reynolds JA, Denlinger DL, et al. A de novo transcriptome of the Asian tiger mosquito, Aedes albopictus, to identify candidate transcripts for diapause preparation[J]. BMC Genomics, 2011, 12: 619. DOI:10.1186/1471-2164-12-619 |