 2021, Vol. 32
2021, Vol. 32扩展功能
文章信息
- 许雪莲, 韩阿祥, 叶诗晴, 关万春, 楼永良
- XU Xue-lian, HAN A-xiang, YE Shi-qing, GUAN Wan-chun, LOU Yong-liang
- 温州口岸截获蜱体内微生物群落结构、抗生素抗性基因及毒力因子的宏基因组分析
- Metagenomic analysis of microbial community structure, antibiotic resistance genes, and virulence factors of ticks captured at Wenzhou port, Zhejiang province, China
- 中国媒介生物学及控制杂志, 2021, 32(6): 763-771
- Chin J Vector Biol & Control, 2021, 32(6): 763-771
- 10.11853/j.issn.1003.8280.2021.06.021
-
文章历史
- 收稿日期: 2021-05-11




蜱是一类可在脊椎动物(包括人类)体表寄生并吸血的节肢动物,在我国分布广泛,种群数量大,可通过叮咬将携带的病原体传播给人类,从而引起多种人畜共患病。目前已报道的由蜱叮咬或直接传播导致的人畜共患病中超过16种与人类相关,19种与动物相关,主要包括蜱传脑炎[1]、莱姆病[2]、人粒细胞无形体病[3]、Q热[4]、发热伴血小板减少综合征[5]等。同时蜱可以通过人员流动、货物运输、动物引进等途径跨境传入[6]。近年来,我国口岸的进口动物皮毛、皮张中多次被检出亚洲璃眼蜱(Hyalomma asiaticum asiaticum)、盾陷璃眼蜱(Hy. excavatum)、绚丽花蜱(Amblyomma pomposum)以及囊形扇头蜱(Rhipicephalus bursa)[7-10]。蜱传病原体已严重威胁人和动物健康。
病原菌之所以能侵入宿主并在其体内繁殖,是一系列毒力因子之间相互协调作用的结果。细菌毒力因子通常是使病原菌能够寄生于宿主的蛋白质,包括与黏附,侵袭,分泌系统,毒素和摄铁系统相关的基因产物[11]。这些毒力因子参与细菌的黏附、侵袭、逃避宿主免疫防御,使得其能在宿主细胞内生存、复制及形成和维持生物被膜[12-13]。例如外毒素,可以损坏细胞膜从而作用于宿主细胞表面,也可以通过与细胞内靶标结合,阻断必需的细胞生物学过程,如翻译、信号转导、细胞内运输和肌动蛋白细胞骨架重排[14];肺炎链球菌的荚膜结构可在空间上对抗体与同源细胞表面抗原的结合过程进行阻断,导致宿主体内的吞噬作用和补体介导的裂解降低[15]。研究此类病原菌的毒力因子,有利于了解病原菌和宿主之间的相互作用。
此外,病原菌的耐药性也是一个值得关注的问题。抗生素作为20世纪最重要的医学发现之一,在控制感染性方面具有重要作用,然而,由于近年来抗生素的滥用,导致了一大批耐药性致病菌的出现。已有研究表明,在饲料中加入金霉素进行喂养的肉牛粪便中大肠埃希菌和肠球菌的耐药率相对于对照显著增加[16]。同时,耐药基因的水平转移特性使耐药性在不同细菌种属间传播,加速了耐药水平的提高。蜱作为专性吸血的体外寄生动物,在传播多种病原体导致疾病的同时还可以通过血餐进一步传播抗生素抗性基因(antibiotic resistance genes,ARGs),增加相关疾病治疗难度。因此,了解蜱携带的病原体及其ARGs、毒力因子,对于了解蜱传疾病致病机制,预防蜱传疾病的暴发及有效治疗具有重要意义。
微生物群落通常由数百种微生物组成[17],目前已有的蜱相关微生物群落有限分析表明,其复杂程度高,人类病原体仅占其中很小的组成部分[18],因此选择一次测定即可同时完成多个物种鉴定的方法在蜱的微生物多样性研究中具有很大优势。随着宏基因组学的兴起和近年来二代测序技术的迅速发展,实现了对上百份样本同时进行高通量核酸分子测序,该方法不依赖于细菌的分离培养,尤其适合从混杂的环境样本或临床样本中发现重要的病原微生物,成为鉴定未知病原菌较为有效的技术手段[19]。因此,本研究采用宏基因组测序的方法,对温州口岸截获的蜱进行微生物群落结构、ARGs及毒力因子分析,结合生物信息学分析手段,以期了解由进口贸易跨境传入的蜱可能传播的疫病,以达到预警及有效防治的目的。
1 材料与方法 1.1 样本采集于2019年从温州海关获取一批蜱样本,随机挑选10只,分成5组,分别编号A1~A5。该批蜱由温州检验检疫局从一批进口南非盐湿牛皮上截获,经鉴定为无色扇头蜱(Rh. decoloratus),并于-80 ℃进行保存。
1.2 DNA提取首先对收集的蜱样本进行消毒处理,经75%乙醇溶液浸泡3次,每次10 min,用双蒸水溶液反复清洗3次,风干。将消毒后的蜱置于无菌研钵中用无菌杵在液氮中粉碎后,将全蜱匀浆置于1.5 ml的Eppendoff无菌试管中,悬浮在1 ml无菌盐水中。经400× g离心5 min,将含有蜱DNA的上清液转移到新的无菌试管中。基因组DNA由广东美格基因科技有限公司使用HiPure Bacterial DNA Kits(Magen,广州)按照使用操作说明书进行提取。
1.3 文库构建及高通量测序提取的DNA样本经1%琼脂糖凝胶电泳检测DNA的完整性和纯度,并用Qubit 3.0(ThermoFisherScience,Waltham,USA)和NanodropOne(Thermo Fisher Scientific,Waltham,USA)同时测定DNA浓度和纯度。检测合格的DNA样品经超声随机打断,将打断后得到的短片段DNA使用alfa-seq DNA library prep kit_manual_cn-final试剂盒(Magen,广州)进行文库构建(按照使用操作说明书),利用Qsep 400(Bioptic,中国台湾)对建好的文库进行质检,对于质检合格的文库采用Novaseq 6000高通量测序平台进行PE150测序。
1.4 生物信息学分析为保证后续分析结果的可信度,使用Trimmomactic软件对原始数据进行质控;利用MEGAHIT软件(https://github.com/voutcn/megahit)对质控后的Clean data进行序列组装;采用Prodigal软件进行ORF预测;采用Linclust软件(https://www.nature.com/articles/s41467-018-04964-5)进行基因聚类并去冗余,获得非冗余的gene catalogue(Unigenes);通过BLASTP(V2.2.31+,http://blast.ncbi.nlm.nih.gov/Blast.cgi)软件进行物种及功能注释;通过主坐标分析(principal coordinate analysis,PCoA)进行细菌群落结构的比较分析(β-多样性),比较样品间的差异情况。
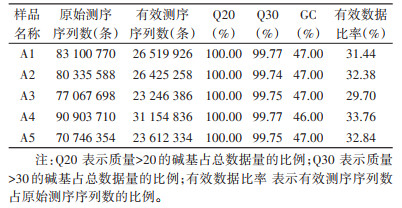
2 结果 2.1 宏基因组测序质控本次测序共获得402 154 120条原始读长,经质控过滤,有效数据中测序错误率小于1.00%(Q20)和0.10%(Q30)的碱基数目比例分别达到100%和99.00%以上,显示测序数据可靠性较高(表 1)。基因预测共得到765 655条开放阅读框(ORFs)用于后续物种注释和基因功能分析。
|
通过与美国国家生物信息中心蛋白数据库NCBI-NR数据库比对,本次测序共注释到35个门类,9个最丰富的门在图 1中显示,依次为厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、广古菌门(Euryarchaeota)、拟杆菌门(Bacteroidetes)、放线菌门(Actinobacteria)、蓝菌门(Cyanobacteria)、疣微菌门(Verrucomicrobia)、衣原体门(Chlamydiae)、浮霉菌门(Planctomycetes)。在5组样本中,厚壁菌门均是优势菌门,分别占总体的70.87%、68.41%、69.77%、72.28%、66.16%。在属水平上,共注释到634个菌属,9个丰度较高的属见图 2,分别是链球菌属、芽孢杆菌属、肠杆菌属、真杆菌属、嗜盐杆菌属、Lachnoclostridium、海杆菌属、副痘病毒属、沙门菌属。其中以链球菌属丰度最高,分别占样本总菌属的57.58%、56.41%、56.38%、57.94%、55.31%,芽孢杆菌属次之,分别占12.32%、12.45%、12.13%、11.28%、11.21%。在种水平上,共注释到1 308个种,其中包括一些常见的条件致病菌(图 3):变形链球菌、蜡样芽孢杆菌、肠沙门菌、肺炎链球菌,共占注释总水平的71.00%以上。其中变形链球菌丰度最高,分别占总体的56.59%、55.87%、57.55%、59.09%、55.57%,蜡样芽孢杆菌次之,为13.03%、13.21%、13.28%、12.38%、12.12%。

|
| 图 1 无色扇头蜱在门水平上的微生物群落结构 Figure 1 Microbial community structure of samples of Rhipicephalus decoloratus at phylum level |
| |

|
| 图 2 无色扇头蜱在属水平上的微生物群落结构 Figure 2 Microbial community structure of samples of Rhipicephalus decoloratus at genus level |
| |

|
| 图 3 无色扇头蜱在种水平上的微生物群落结构 Figure 3 Microbial community structure of samples of Rhipicephalus decoloratus at species level |
| |
根据多样本物种、功能和基因数目比较的结果显示(图 4),5组样品共有基因数高达488 593个(图 4A),A1~A5样品独有的基因数分别为12 794、28 761、24 991、35 068、24 845,相比之下A4蜱样本中微生物独有的基因数最多。对属水平上的物种数目进行分析,5个样品共有500个属(图 4B),其中A1、A4、A5各独有1个属,A2独有3个属,A3独有2个属。此外,5组样品共有Kyoto encyclopedia of genus and genomes(KEGG)(Lever 2)功能数目47个(图 4C)。使用PCoA方法展示本次5组蜱样本微生物群落结构的差异程度(图 5),结果显示5个样本间的微生物组成相似,其中第1轴(axis 1)贡献度为58.80%,第二轴(axis 2)贡献度为31.60%。

|
| 注:A基因数目韦恩图;B属水平上的物种数目韦恩图;C KEGG功能数目韦恩图。 图 4 无色扇头蜱多样本比较韦恩图 Figure 4 Venn diagram of multi-sample comparison of Rhipicephalus decoloratus |
| |

|
| 注:PCoA指主坐标分析;method=bray指计算距离矩阵的方法为相异度(Bray-Curtis)距离;Species表示基于物种水平分析。 图 5 无色扇头蜱基于物种水平的主坐标分析 Figure 5 Principal co-ordinates analysis based on species level of Rhipicephalus decoloratus |
| |
将非冗余基因集序列与KEGG的基因数据库进行基因的同源性比对(设定阈值为e-value≤0.001),获得基因注释信息,结果见图 6。本次研究中蜱所携带微生物在数据库中注释到数量最多的通路是新陈代谢,其次是遗传信息处理、环境信息处理、细胞过程、人类疾病、有机体系统。其中人类疾病相关通路注释到的基因中585个基因与癌症通路相关,41个基因与免疫疾病代谢通路相关,277个基因与神经退行性疾病通路相关,238个基因与内分泌代谢疾病相关,126个基因与心血管疾病相关,1 085个基因与传染病相关,567个基因与耐药性通路相关。

|
| 图 6 无色扇头蜱KEGG通路注释 Figure 6 KEGG pathway annotation of Rhipicephalus decoloratus |
| |
本研究通过与抗性基因数据库(antibiotic resistance genes database,ARDB)数据库比对,注释到16种共53个抗生素抗性基因(图 7、表 2)。样本中主要的耐药基因是Acr,其次是bacA,分别占30.19%和20.75%。注释到的16种抗性基因中,其中bacA、bcr_mfs与杆菌肽耐药相关,Acr与大环内酯类、氨基糖苷类、β-内酰胺酶、甘氨酰环素、吖啶黄素多种药物耐药相关,arnA与对粘菌素耐药相关,bla_c与头孢霉菌素耐药相关,fos、rosab与磷霉素耐药相关,ksgA与春雷霉素耐药相关,macAB与大环内酯类耐药相关,mdtG、mdtH、mdtL与磷霉素耐药相关,mdtK与依诺沙星、诺氟沙星耐药相关,tetC基因与四环素类抗生素耐药相关。此外,对于不同基因耐药机制进行分析发现(表 2),Acr、bcr_mfs、emrD、macAB、mdtG、mdtH、mdtK、mdtL、rosab、tetC等10种基因参与外排泵作用而介导耐药,bacA、arnA、bla_c、fos、ksgA、mdfA等6种基因分别参与二甲基烯丙基顺转移酶、核苷二磷酸糖差向异构酶、β-内酰胺酶、谷胱甘肽转移酶、16S rRNA的甲基化酶、磷脂丝胺酸转位酶等而介导细菌耐药性。

|
| 图 7 无色扇头蜱所有抗性基因基因数目 Figure 7 Number of antibiotic resistance genes of Rhipicephalus decoloratus |
| |

|
通过与细菌毒力因子数据库(virulence factor database,VFDB)比对,共3 923个基因获得毒力基因注释,预测获得的基因注释结果见图 8。探索了4种类型的毒力因子,包括防御型毒力因子、毒力相关基因的调控、非特异性毒力因子和进攻型毒力因子。进攻型毒力因子主要与黏附、分泌系统、侵袭、内毒素、宿主免疫逃避、细菌毒素相关。非特异性毒力因子与铁吸收系统、镁吸收系统相关。防御型毒力因子主要与位相变异、血清抵抗、抗吞噬作用、应激蛋白、酶、补体蛋白酶、Ig蛋白酶相关。在16种毒力因子中与黏附有关的基因最多(22.66%),其次为与分泌系统(13.97%)、侵袭(13.56%)、铁吸收系统(10.86%)、位相变异(10.60%)、内毒素(9.07%)有关的基因。此外,还注释到了与血清抵抗(5.25%)、调控(3.93%)、抗吞噬作用(3.85%)、应激蛋白(3.64%)以及少量与宿主免疫逃避(0.92%)、细菌毒素(0.89%)、镁吸收系统(0.59%)、胞外酶(0.10%)、补体蛋白酶(0.08%)、Ig蛋白酶(0.03%)相关的基因。

|
| 图 8 无色扇头蜱所有毒力因子的基因数目 Figure 8 Number of all virulence genes of Rhipicephalus decoloratus |
| |
蜱为人、家畜及野生动物的体外寄生虫,不仅可咬伤皮肤,而且是螺旋体、立克次体、病毒、细菌等多种病原体的媒介[20]。蜱体内微生物群落结构复杂,仅以常规的培养分离等手段进行检疫,无法全面了解蜱疫情。因此,本研究应用宏基因组学技术对温州口岸在进口南非盐湿牛皮上检疫发现的无色扇头蜱体内微生物群落结构特征,及抗生素抗性基因、毒力因子携带情况进行研究,发现该5份蜱样本微生物群落结构相似,在门水平上均以厚壁菌门、变形菌门为主要菌门,该结果与Xiang等[21]的研究结果相符。在属水平上5份样本均以链球菌属、芽孢杆菌属、盐杆菌属为主要菌属。Chiuya等[22]在关于肯尼亚牲畜市场及屠宰场的蜱传病原体研究中检测到无色扇头蜱携带立克次体、无形体等病原体。此外,在对新疆维吾尔自治区家畜蜱中和韩国蜱传病原体的研究中也检测到潜在的蜱传病原体——立克次体属[23-24]。立克次体是一大类严格细胞内寄生的原核细胞型微生物,以节肢动物(主要为蜱)作为寄生宿主及载体[25],其可在蜱的所有器官中繁殖,尤其是唾液腺,因此可在血餐过程中传播给动物宿主。本研究中未注释到相应菌属,可能与物种及其宿主特异性,地理环境,植物覆盖,季节和采样源等条件有关。尽管未注释到特定的蜱传病原体,但在种水平上,我们注释到一些常见的条件致病菌(图 3),主要包括:变形链球菌、蜡样芽孢杆菌、肠沙门菌等。其中变形链球菌是引起龋病最主要的致龋菌,同时其被证明与许多全身系统性疾病有密切联系。肠炎沙门菌属于无宿主特异性而有侵害性的病原菌之一,该菌不仅能引起家禽发病死亡造成严重的经济损失,而且可通过被污染的家禽产品进行传播,危害人类健康。近年来,我国发生的沙门菌病中大部分由肠炎沙门菌引起[26]。本研究中蜱体内的肠炎沙门菌可能来源于无色扇头蜱的宿主或其生活环境中,并可能经污染禽畜类食品通过食物链进行传播。此外,蜡状芽孢杆菌主要引起食物中毒和严重眼部感染[27],也有报道称蜡状芽孢杆菌对β-内酰胺和碳青霉烯有抗药性[28]。若宿主携带有此类耐药病原体,人类可能通过食用动物源性食品或与患病动物接触而感染发病。因此,在针对口岸截获蜱检疫中,我们不仅要关注特定的蜱传病原体,还需要关注其携带的其他病原微生物,加强对蜱的监测,有效做好蜱媒传染病的防控工作。
在病原菌致病过程中多种毒力因子参与并发挥其特定功能,这些基因编码一系列与宿主细胞直接作用的分子,如毒素、胞外酶、分泌系统、生物膜形成蛋白、铁载体以及具有抗吞噬特性的多糖[29],从而起到迁移定植,攻击宿主防御机制,抗吞噬等作用[11]。因此,本研究首次关注蜱体内微生物毒力因子,发现此次5份蜱样本中的毒力因子主要与黏附、分泌系统、侵袭、铁吸收系统相关。细菌黏附素可分为非菌毛黏附素(afimbrial adhesin)和菌毛黏附素(fimbrial adhesin)两种。菌毛有助于菌体黏附到宿主细胞表面,在感染中起启动作用。以往的研究表明,带菌毛菌株的致病性及活性均比先天无菌毛菌株或菌毛缺失的菌株强。分泌系统作为重要的毒力因子分泌途径,在细菌的致病性方面发挥重要作用[30]。例如气溶素、蛋白酶、溶血素等能够破坏宿主细胞,造成组织损伤和坏死,这些毒力因子的分泌都需要Ⅱ型分泌系统(T2SS)参与;同时,Ⅱ型分泌系统对细菌的运动能力及生物膜形成等也具有重要意义[31]。目前,已在革兰阴性菌中发现9种不同的分泌系统(Ⅰ~Ⅸ型)[32]。通过分泌系统病原微生物可直接将其产生的特异蛋白导入宿主细胞,从而引起感染。铁是大多数生物生长必需的元素,有研究表明,细菌摄铁系统获得铁的效率的提高是细菌毒力增加的主要因素,细菌致病性跟细菌摄取铁的能力有很强的关联[33]。例如鼠疫耶尔森菌(Yersinia pestis)体内具有高效能的摄铁系统,该菌种对铁的高效率获取有助于其在机体内定植并进一步增殖导致宿主感染[34]。由此可见,铁转运系统虽未直接涉及致病过程,但对细菌致病性的产生也至关重要。除上述3种毒力因子外,我们还注释到毒素、Ig蛋白酶等多种毒力因子。针对细菌毒力因子的研究,有利于后续疫苗开发,该技术通过中和毒力因子达到抗感染目的,其不仅阻止了病原菌的致病过程,也避免了选择压力,从而降低了细菌的耐药率。但鉴于细菌毒力系统的复杂性,对毒力因子的分子机制以及病原菌与宿主的相互作用还需进一步了解。
此外,蜱中病原体除可引起多种人畜共患病外,还可作为潜在抗性基因贮藏库,通过特定的方式向人类传播抗生素耐药性。关于ARGs,通过与ARDB数据库比对,在本研究中共注释到16种抗性基因,主要为Acr和bacA,其分别与大环内酯类、氨基糖苷类、β-内酰胺酶、甘氨酰环素、吖啶黄素多种药物耐药及杆菌肽耐药相关。现已描述的细菌对抗生素产生抗性的机制包括代谢抗生素的酶的产生、与抗生素修饰相关的酶的产生、阻止其与细胞相应位点结合的抗生素靶标的修饰和外排泵系统[35-38]。在本研究中,对这16种抗性基因耐药机制进行分析,发现其抗性主要由多药外排泵介导(表 2)。细菌外排泵是细菌细胞膜上的一类蛋白,能够通过膜通道蛋白外排多种抗菌药物,阻止药物在细菌体内聚集,从而导致致病菌对大环内酯类、氨基糖苷类、β-内酰胺酶、甘氨酰环素、吖啶黄素、磷霉素等多种药物耐药。因此,在本研究中注释到相对高丰度的多药耐药外排泵并不奇怪。蜱是以吸血为生的体外寄生虫,大量寄生于动物体表,因此本研究中的ARGs可能主要来源于蜱的寄生宿主。有研究通过对成年奶牛、小牛犊和断奶前犊牛粪便进行宏基因组测序,发现在动物的粪便中含有大量的ARGs[39],并且通过宏基因组测序发现许多ARGs为动物、人类粪便所共有,说明ARGs可能通过某些途径如食物链等从动物相关细菌传播给人类。其中,Kastner等[40]在对多类畜禽类发酵制品的检测中,发现与四环素类、链霉素类、β-内酰胺类相关的抗生素抗性基因。Garofalo等[41]在意大利的家禽肉及香肠制品中也注释到四环素类、β-内酰胺类等多种抗性基因。这些研究表明,ARGs广泛存在于畜禽类食品中,并可能部分通过禽畜肉类或奶制品进行传递。另一方面,这些抗性基因可能经动物粪便排放,进入土壤及水体环境中,最终进入果蔬类食品,以食品作为ARGs的载体,将极大威胁到人类健康,增加相关疾病的治疗难度。因此,针对蜱及其宿主体内的抗性基因的种类及分布情况需要引起广泛重视。ARGs的分布受多种因素影响,除环境因素影响外,袁立霞等[42]研究制药废水厂发现少数菌属与抗性基因存在相关性,说明抗性基因的分布与相关特定菌属有关。因此,可以通过控制相关菌属的丰度及其他条件来消减抗性基因丰度。
本研究利用宏基因组技术手段,对难培养或不可培养的微生物进行鉴定,为进一步了解我国入境皮革携带的蜱样本体内的微生物提供了一种新型有效的检验检疫手段。测序结果发现许多未被分类的微生物,说明蜱中微生物多样性可能比我们先前推测的要大得多,验证了通过高通量测序技术能更全面了解生物学信息,从而有利于发现和认识一些新的微生物。同时,在本研究的蜱体内检出变形链球菌、蜡样芽孢杆菌、肠沙门菌等条件致病菌,该结果提示相关机构需加强对于入境皮革及动物的检验检疫工作,畜牧业从业人员及野外工作者等应该做好自我防护以切断传播途径。此外,对蜱体内微生物毒力因子和病原体抗生素抗性基因的鉴定,有助于更好地理解相关细菌致病机理及其与宿主的相互作用,指导临床用药,改善治疗效果。但以现有的宏基因组学技术手段,仍有许多存在但未被发现的菌种,对菌群的研究仍然存在一定局限性。同时,本研究中仅选取温州检验检疫局在同一批进口南非盐湿牛皮上截获的无色扇头蜱为研究对象,样本种类及样本数量较少。因此,后续可以扩大样本的种类、来源及数量,进一步开展口岸截获蜱相关微生物群落结构等研究,以达到全面了解入境蜱体内病原体信息,为我国蜱媒传染病的有效防控及治疗提供参考依据的目的。
志谢 温州出入境检验检疫局王素华博士在取样方面给予大力帮助,特此志谢利益冲突 无
| [1] |
Shi JM, Hu ZH, Deng F, et al. Tick-borne viruses[J]. Virol Sin, 2018, 33(1): 21-43. DOI:10.1007/s12250-018-0019-0 |
| [2] |
Koedel U, Fingerle V, Pfister HW. Lyme neuroborreliosis-epidemiology, diagnosis and management[J]. Nat Rev Neurol, 2015, 11(8): 446-456. DOI:10.1038/nrneurol.2015.121 |
| [3] |
Ismail N, McBride JW. Tick-borne emerging infections: ehrlichiosis and anaplasmosis[J]. Clin Lab Med, 2017, 37(2): 317-340. DOI:10.1016/j.cll.2017.01.006 |
| [4] |
Chmielewski T, Tylewska-Wierzbanowska S. Q fever at the turn of the century[J]. Pol J Microbiol, 2012, 61(2): 81-93. DOI:10.33073/pjm-2012-011 |
| [5] |
Zeng PB, Yang ZD, Bakkour S, et al. Development and validation of a real-time reverse transcriptase PCR assay for sensitive detection of SFTSV[J]. J Med Virol, 2017, 89(7): 1131-1138. DOI:10.1002/jmv.24760 |
| [6] |
赵丹云, 孙毅, 陈卫军, 等. 天津口岸首次截获随人入境外来蜱的形态学及分子鉴定[J]. 中国国境卫生检疫杂志, 2016, 39(4): 256-259. Zhao DY, Sun Y, Chen WJ, et al. Morphology and molecular identification of an imported tick with entry passenger intercepted at Tianjin port[J]. Chin J Front Health Quar, 2016, 39(4): 256-259. DOI:10.16408/j.1004-9770.2016.04.009 |
| [7] |
余招锋, 徐慧, 陈鹏程, 等. 一批泰国进境板材中截获的囊形扇头蜱的鉴定和检疫[J]. 中国动物检疫, 2017, 34(2): 58-60. Yu ZF, Xu H, Chen PC, et al. Identification and quarantine towards Rhipicephalus bursa in intercepted timbers imported from Thailand[J]. China Anim Health Insp, 2017, 34(2): 58-60. DOI:10.3969/j.issn.1005-944X.2017.02.016 |
| [8] |
吕洁毅, 何振毅, 刘谢, 等. 佛山口岸首次截获绚丽花蜱[J]. 寄生虫与医学昆虫学报, 2016, 23(4): 237-241. Lyu JY, He ZY, Liu X, et al. An exotic species of Amblyomma intercepted at Foshan frontier port[J]. Acta Parasitol Med Entomol Sin, 2016, 23(4): 237-241. DOI:10.3969/j.issn.1005-0507.2016.04.008 |
| [9] |
徐军, 王安东, 戴莉, 等. 中哈边境阿拉山口口岸输入性亚洲璃眼蜱的种类鉴定与分析[J]. 中国兽医杂志, 2016, 52(11): 17-19. Xu J, Wang AD, Dai L, et al. Species identification of an imported tick sample of Hyalomma asiaticum asiaticum by genetic analysis at Alashankou port, China-Kazakhstan border[J]. Chin J Vet Med, 2016, 52(11): 17-19. |
| [10] |
赵爽, 胡文海, 闫清丽, 等. 南沙口岸首次从入境牛皮中截获盾陷璃眼蜱[J]. 中国媒介生物学及控制杂志, 2014, 25(5): 452-454. Zhao S, Hu WH, Yan QL, et al. Investigation of Hyalomma excavatum Koch first captured at Nansha port, Guangzhou, China[J]. Chin J Vector Biol Control, 2014, 25(5): 452-454. DOI:10.11853/j.issn.1003.4692.2014.05.017 |
| [11] |
Chen LH, Xiong ZH, Sun LL, et al. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors[J]. Nucleic Acids Res, 2012, 40(D1): D641-D645. DOI:10.1093/nar/gkr989 |
| [12] |
Mühlen S, Dersch P. Anti-virulence strategies to target bacterial infections[M]//Stadler M, Dersch P. How to overcome the antibiotic crisis. Cham: Springer, 2015: 147-183. DOI: 10.1007/82_2015_490.
|
| [13] |
Ruer S, Pinotsis N, Steadman D, et al. Virulence-targeted antibacterials: concept, promise, and susceptibility to resistance mechanisms[J]. Chem Biol Drug Des, 2015, 86(4): 379-399. DOI:10.1111/cbdd.12517 |
| [14] |
Harms A, Brodersen DE, Mitarai N, et al. Toxins, targets, and triggers: an overview of toxin-antitoxin biology[J]. Mol Cell, 2018, 70(5): 768-784. DOI:10.1016/j.molcel.2018.01.003 |
| [15] |
Yother J. Capsules of Streptococcus pneumoniae and other bacteria: paradigms for polysaccharide biosynthesis and regulation[J]. Annu Rev Microbiol, 2011, 65: 563-581. DOI:10.1146/annurev.micro.62.081307.162944 |
| [16] |
Platt TM, Loneragan GH, Scott HM, et al. Antimicrobial susceptibility of enteric bacteria recovered from feedlot cattle administered chlortetracycline in feed[J]. Am J Vet Res, 2008, 69(8): 988-996. DOI:10.2460/ajvr.69.8.988 |
| [17] |
Kim Y, Koh I, Rho M. Deciphering the human microbiome using next-generation sequencing data and bioinformatics approaches[J]. Methods, 2015, 79-80: 52-59. DOI:10.1016/j.ymeth.2014.10.022 |
| [18] |
Martello LA, Wadgaonkar R, Gupta R, et al. Characterization of Trypanosoma cruzi infectivity, proliferation, and cytokine patterns in gut and pancreatic epithelial cells maintained in vitro[J]. Parasitol Res, 2013, 112(12): 4177-4183. DOI:10.1007/s00436-013-3609-7 |
| [19] |
Manichanh C, Chapple CE, Frangeul L, et al. A comparison of random sequence reads versus 16S rDNA sequences for estimating the biodiversity of a metagenomic library[J]. Nucleic Acids Res, 2008, 36(16): 5180-5188. DOI:10.1093/nar/gkn496 |
| [20] |
Boulanger N, Boyer P, Talagrand-Reboul E, et al. Ticks Tick Borne Dis[J]. Méd Mal Infect, 2019, 49(2): 87-97. DOI:10.1016/j.medmal.2019.01.007 |
| [21] |
Xiang LL, Poźniak B, Cheng TY. Bacteriological analysis of saliva from partially or fully engorged female adult Rhipicephalus microplus by next-generation sequencing[J]. Antonie van Leeuwenhoek, 2017, 110(1): 105-113. DOI:10.1007/s10482-016-0780-8 |
| [22] |
Chiuya T, Masiga DK, Falzon LC, et al. Tick-borne pathogens, including Crimean-Congo haemorrhagic fever virus, at livestock markets and slaughterhouses in western Kenya[J]. Transbound Emerg Dis, 2021, 68(4): 2429-2445. DOI:10.1111/tbed.13911 |
| [23] |
Li YC, Wen XX, Li M, et al. Molecular detection of tick-borne pathogens harbored by ticks collected from livestock in the Xinjiang Uygur Autonomous Region, China[J]. Ticks Tick-Borne Dis, 2020, 11(5): 101478. DOI:10.1016/j.ttbdis.2020.101478 |
| [24] |
Jiang J, Choi YJ, Kim J, et al. Distribution of Rickettsia spp. in ticks from northwestern and southwestern provinces, Republic of Korea[J]. Korean J Parasitol, 2019, 57(2): 161-166. DOI:10.3347/kjp.2019.57.2.161 |
| [25] |
Merhej V, Angelakis E, Socolovschi C, et al. Genotyping, evolution and epidemiological findings of Rickettsia species[J]. Infect Genet Evol, 2014, 25: 122-137. DOI:10.1016/j.meegid.2014.03.014 |
| [26] |
魏琼, 刘翔, 张燕飞, 等. 宁夏地区食源性与人源沙门菌耐药性与血清型对比研究[J]. 中国抗生素杂志, 2016, 41(9): 707-709, 717. Wei Q, Liu X, Zhang YF, et al. Serotype distribution and drug resistance of food-born and human orign Salmonella in Ningxia[J]. Chin J Antibiot, 2016, 41(9): 707-709, 717. DOI:10.3969/j.issn.1001-8689.2016.09.012 |
| [27] |
Bottone EJ. Bacillus cereus, a volatile human pathogen[J]. Clin Microbiol Rev, 2010, 23(2): 382-398. DOI:10.1128/CMR.00073-09 |
| [28] |
Torkar KG, Bedenić B. Antimicrobial susceptibility and characterization of metallo-β-lactamases, extended-spectrum β-lactamases, and carbapenemases of Bacillus cereus isolates[J]. Microb Pathog, 2018, 118: 140-145. DOI:10.1016/j.micpath.2018.03.026 |
| [29] |
Wu HJ, Wang AHJ, Jennings MP. Discovery of virulence factors of pathogenic bacteria[J]. Curr Opin Chem Biol, 2008, 12(1): 93-101. DOI:10.1016/j.cbpa.2008.01.023 |
| [30] |
尹磊, 祁克宗, 宋祥军, 等. 大肠杆菌Ⅲ型分泌系统2毒力岛研究进展[J]. 微生物学通报, 2017, 44(12): 3031-3037. Yin L, Qi KZ, Song XJ, et al. Type Ⅲ secretion system 2 pathogenicity islands of Escherichia coli[J]. Microbiol China, 2017, 44(12): 3031-3037. DOI:10.13344/j.microbiol.china.170468 |
| [31] |
Francetic O, Belin D, Badaut C, et al. Expression of the endogenous type Ⅱ secretion pathway in Escherichia coli leads to chitinase secretion[J]. EMBO J, 2000, 19(24): 6697-6703. DOI:10.1093/emboj/19.24.6697 |
| [32] |
余乐正, 柳凤娟, 鄢南南, 等. 革兰阴性菌分泌系统及其分泌产物研究进展[J]. 动物医学进展, 2017, 38(8): 80-84. Yu LZ, Liu FJ, Yan NN, et al. Progress on secretion systems and secretory products of gram-negative bacteria[J]. Prog Vet Med, 2017, 38(8): 80-84. DOI:10.16437/j.cnki.1007-5038.2017.08.017 |
| [33] |
Shon AS, Bajwa RPS, Russo TA. Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed[J]. Virulence, 2013, 4(2): 107-118. DOI:10.4161/viru.22718 |
| [34] |
Osorio CR, Juiz-Río S, Lemos ML. A siderophore biosynthesis gene cluster from the fish pathogen Photobacterium damselae subsp. piscicida is structurally and functionally related to the Yersinia high-pathogenicity island[J]. Microbiology (Reading),, 2006, 152(Pt 11): 3327-3341. DOI:10.1099/mic.0.29190-0 |
| [35] |
Blair JMA, Webber MA, Baylay AJ, et al. Molecular mechanisms of antibiotic resistance[J]. Nat Rev Microbiol, 2015, 13(1): 42-51. DOI:10.1038/nrmicro3380 |
| [36] |
Fournier PE, Richet H, Weinstein RA. The epidemiology and control of Acinetobacter baumannii in health care facilities[J]. Clin Infect Dis, 2006, 42(5): 692-699. DOI:10.1086/500202 |
| [37] |
Kempf M, Rolain JM. Emergence of resistance to carbapenems in Acinetobacter baumannii in Europe: clinical impact and therapeutic options[J]. Int J Antimicrob Agents, 2012, 39(2): 105-114. DOI:10.1016/j.ijantimicag.2011.10.004 |
| [38] |
Vila J, Martí S, Sánchez-Céspedes J. Porins, efflux pumps and multidrug resistance in Acinetobacter baumannii[J]. J Antimicrob Chemother, 2007, 59(6): 1210-1215. DOI:10.1093/jac/dkl509 |
| [39] |
Haley BJ, Kim SW, Cao H, et al. Genomic and metagenomic analysis of antibiotic resistance in dairy animals[J]. J Anim Sci, 2017, 95(Suppl 4): S131. DOI:10.2527/asasann.2017.265 |
| [40] |
Kastner S, Perreten V, Bleuler H, et al. Antibiotic susceptibility patterns and resistance genes of starter cultures and probiotic bacteria used in food[J]. Syst Appl Microbiol, 2006, 29(2): 145-155. DOI:10.1016/j.syapm.2005.07.009 |
| [41] |
Garofalo C, Vignaroli C, Zandri G, et al. Direct detection of antibiotic resistance genes in specimens of chicken and pork meat[J]. Int J Food Microbiol, 2007, 113(1): 75-83. DOI:10.1016/j.ijfoodmicro.2006.07.015 |
| [42] |
袁立霞, 罗晓, 张文丽, 等. 制药废水厂微生物群落和多种抗性基因相关性分析[J]. 河北科技大学学报, 2019, 40(2): 175-181. Yuan LX, Luo X, Zhang WL, et al. Correlation between antibiotic resistance genes and microbial communities in pharmaceutical wastewater[J]. J Hebei Univ Sci Technol, 2019, 40(2): 175-181. DOI:10.7535/hbkd.2019yx02012 |