 2016, Vol. 33
2016, Vol. 33 



环加成反应是有机合成中最重要的反应之一,它实现由简单易得的反应物构建复杂的环状结构,在天然产物的全合成中有着广泛应用[1].过渡金属催化环加成反应是近年来发展起来的实现此类反应的新合成方法,极大地丰富了传统环加成反应(如Diels-Alder反应)[2].在过渡金属催化的环加成反应中,反应物通常是含C(sp2)—H键的不饱和化合物,通过直接活化C—H键来实现C—C键的偶联成环是一种原子经济、合成步骤经济的环境友好合成方法[3-4].如何选择性地控制C—H键活化是C—H键官能团化的关键. 利用邻近杂原子基团(如羟基、羧基、酰胺等) 作为导向基团来控制C—H健活化的选择性是目前被广泛采用的方法[5-7].因此该类反应通常得杂环产物而非碳环[8-9].另外,基于(3 + 2)[10-11] 或 (4 + 2)[12-13]的环加成反应通常得到五元或六元杂环产物,七元以上杂环化合物的合成还有待进一步研究[14-15].
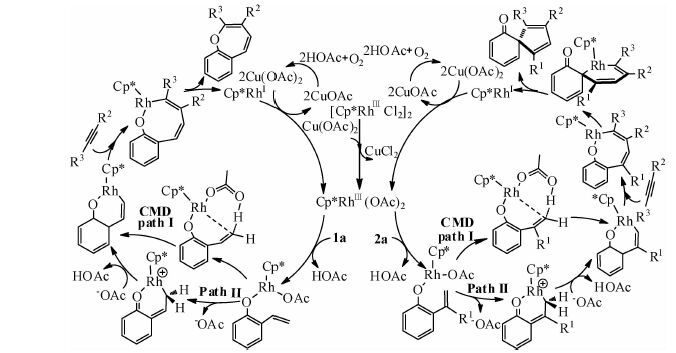
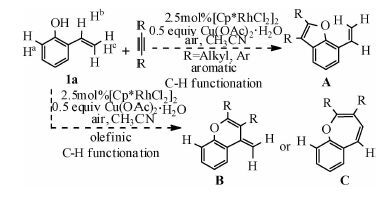
Rh在催化环加成反应中因其对官能团的良好耐受性而被广泛应用[7].Seoane等[16]于2013年报道了使用[Cp*RhCl2]2作为催化剂前驱体,在Cu(OAc)2和O2存在条件下,以—OH为导向基团,催化2-羟基苯乙烯与炔烃进行[5+2]环加成合成七元杂环的反应(eq 1).在2-羟基苯乙烯中有3类C—H键可被活化,对这3类C—H键的活化可分别生成五元、六元及七元含氧杂环,但是该反应仅生成了七元环产物.
近来,该课题组[17]又报道了2-异烯丙基苯酚与炔烃进行[3+2]环加成合成五元螺环的反应(eq 2).这两例反应(eq 1 和 2),使用的催化剂和反应条件相同,反应底物相似,并且均是以羟基为导向基团,但产物完全不同(七元含氧杂环vs.五元螺环),因此这2例反应是过渡金属催化环加成反应中非常少见的具有代表性的反应案例.Seoane 及其合作者对这两例反应的反应机理提出了假设(图 1).但具体的反应机理还不清楚.本文采用密度泛函计算方法对这2例反应的反应机理进行详细的对比研究,以揭示造成这两例反应选择性不同的根源.

|
Download:
|
| 图 1 Seoane 及其合作者提出的反应机理 Fig. 1 Mechanisms proposed by Seoane and co-workers | |
{kind=link}
本文使用B3LYP[18]/BSI方法对所有结构进行优化,这里,BSI代表混合基组,对金属Rh用SDD[19],非金属原子用6-31G(d)基组.通过频率分析计算确定所优化的结构是稳定极小点(无虚频)还是过渡态(仅有一个虚频).然后,使用B3LYP/BSI优化的构型,进一步在M06/BSII方法水平上做考虑溶剂效应的单点校正,BSII代表混合基组,对过渡金属Rh用SDD,非金属原子用6-311++G(d,p).计算中选用实验溶剂(乙腈)和SMD[20]溶剂模型.B3LYP/BSI级别下气相优化的频率分析结果用作298.15 K和1 atm条件下的焓和自由能校正.这种使用M06//B3LYP 结合的计算对有机金属体系研究的可靠性在我们以前许多研究中得到证实[21-22].除非特别说明,文章所有的讨论结果均是采用M06/BSII//B3LYP/BSI级别下计算得到的自由能.所有计算都是用高斯 09软件[23]完成.
2 结果和讨论实验使用多种2-羟基苯乙烯,2-烯基苯酚及炔烃[16-17].为了便于和实验对比,我们选择优化实验条件所用的2-羟基苯乙烯(1a),2-异烯丙基苯酚(2a)和二苯乙炔环加成反应为研究对象.
2.1 [5+2]环加成生成七元含氧杂环的反应 2.1.1 [5+2]环加成生成七元含氧杂环的反应机理实验使用催化量的[Cp*RhCl2]2 催化剂前体及0.5 当量的Cu(OAc)2.以前的实验及计算证明Cp*Rh(OAc)2是该催化条件下产生的活性催化剂[24].因此,本文使用Cp*Rh(OAc)2 作为活性催化剂.在Cp*Rh(OAc)2的结构中,两个羧酸根分别以双齿和单齿形式配位.
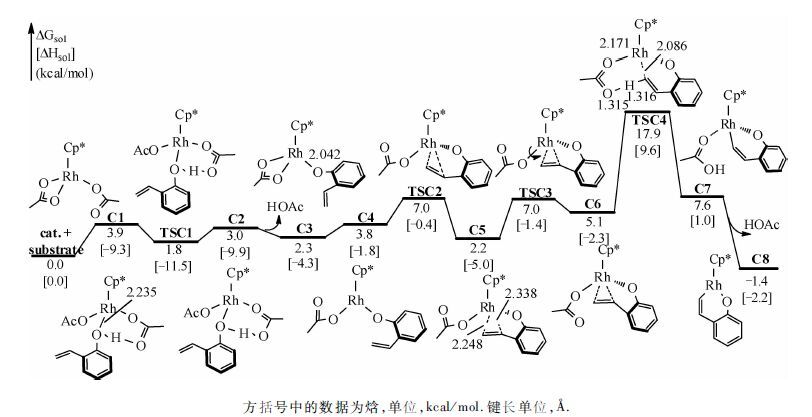
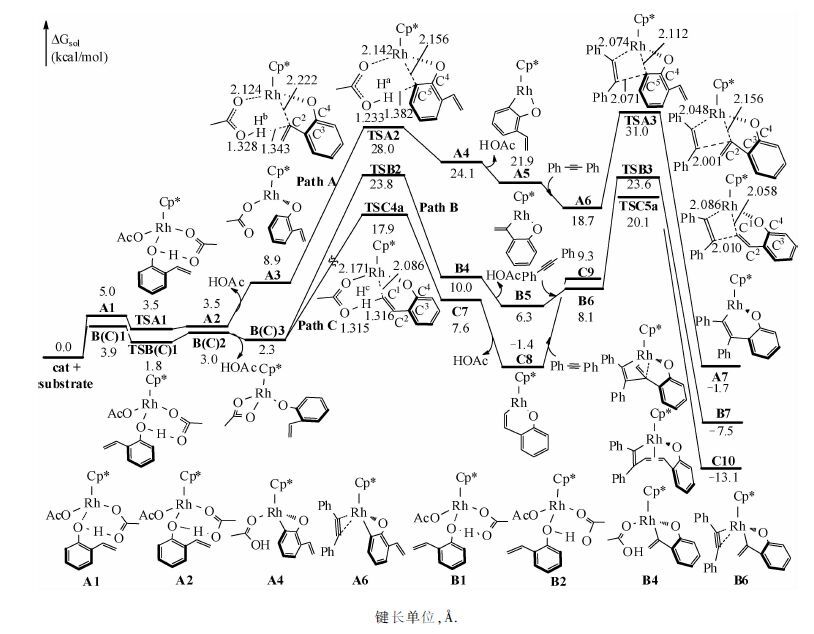
研究结果表明,该反应可分为O—H键断裂、C—H键活化、炔烃迁移插入及还原消除4步.图 2描述前两步反应的反应途径及能量结果.底物1a首先与Rh中心配位,造成Cp*Rh(OAc)2 中的一个双齿羧酸根配体变为单齿,生成C1.在C1中,底物中酚基氧靠近Rh中心,距离为2.235 Å.由于Cp*Rh(OAc)2为18电子稳定结构且位阻较大,该配位不强,虽然体系的焓降低了9.3 kcal/mol,体系的自由能升高了3.9 kcal/mol.C1中酚羟基氢与邻位—OAc的羰基氧间形成氢键,酚羟基氢经TSC1可非常容易地转移到羧酸根配体,形成HOAc,经氢键中间体C2解离,使Rh中心空出一个配位点,单齿羧酸根配体恢复为双齿,生成C3.从C1到C3过程,酚羟基O—Rh配位键转变为共价键(Rh—O间距离为2.042 Å).
从图 2可以看出,TSC1的自由能比C1和C2都低,这是由溶剂效应及热力学校正造成的.若比较三者气相电子能,TSC1的电子能分别比C1和C2高0.4和0.6 kcal/mol.为了便于下一步的乙烯基配位,双齿羧酸根配体再次转变成单齿配体,Rh中心变为三配位,得到C4,失去配位使自由能升高1.5 kcal/mol.C4中,底物乙烯基两端碳与Rh中心的距离分别为3.869 和4.175 Å.之后,经过一个3.2 kcal/mol的位垒(TSC2相对于C4),乙烯基与Rh中心配位生成C5,C5中乙烯基两端碳与Rh中心的距离分别拉近为2.248和2.338 Å.因为在C5中,羧酸根配体的羰基氧的朝向不利于下一步的C—H键活化,因此羧酸根配体绕Rh—O键经TSC3旋转至C6.从C1到C6,最高驻点的相对能量不大于7.0 kcal/mol,最低驻点的相对能量大于零,这些能量结果说明该过程实质上是容易发生的热力学平衡过程.得到C6后,C—H键可以通过两种方式被活化(图 1).一种是传统的CMD(concerted metalation-deprotonation)[25]机理(Path I),在金属中心的协助下,—OAc羰基氧将乙烯基末端氢拔除,实现C—H键活化.另一种是烯基末端碳首先对金属中心进行亲核进攻,生成六元环状中间体,羧酸根配体以阴离子形式解离,然后游离的—OAc将烯基末端氢拔除,实现C—H键的活化(Path II).计算结果表明在这一反应中C—H键的活化是通过CMD机理进行的,即经过协同的六元环状过渡态TSC4,乙烯基末端氢转移给羰基,同时Rh—C形成,生成C8,相对于能量零点,C—H键活化的位垒为17.9 kcal/mol.

|
Download:
|
| 图 2 催化[5+2]环加成反应中O—H键断裂和C—H键活化的反应自由能曲线 Fig. 2 Free energy profiles for the consecutive O—H deprotonation and C—H activation sequences of the Cp*Rh(OAc)2-catalyzed hetero-[5+2] cycloaddition | |
{kind=link}
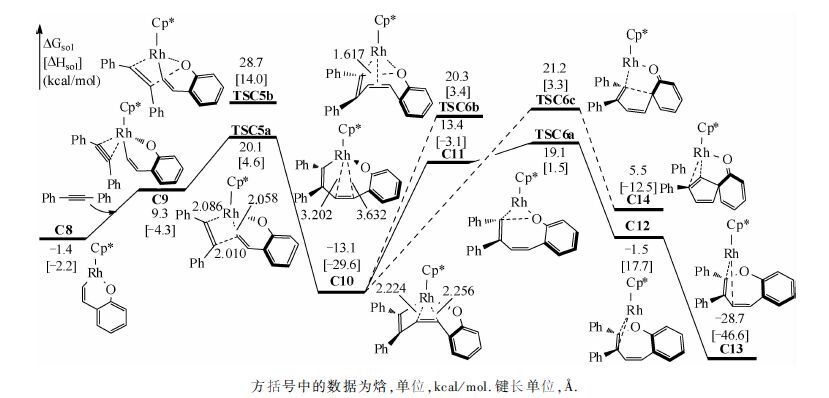
C8是一个16电子不饱和络合物,且金属中心有一个配位点,有利于下一步炔烃的迁移插入.图 3为炔烃迁移插入及还原消除步骤的反应途径.反应物二苯乙炔先与Rh中心配位,自由能升高10.7kcal/mol,得C9.之后,炔烃经过四元环状过渡态(TSC5a)迁移插入至Rh—C键之间,相对于C8,该步位垒为21.5kcal/mol.计算表明炔烃对Rh—O键进行迁移插入的过渡态(TSC5b)自由能比迁移插入至Rh—C键间高8.6kcal/mol,排除该副反应通道.炔烃迁移插入后,得到C10,自由能降低11.7kcal/mol(相对于C8).在C10中,Rh中心与底物中原烯基的双键配位,Rh与双键两端碳的距离分别为2.224 和 2.256 Å,整个分子被扭曲为船式.之后,双键失去配位,得C11,此时,Rh与双键两端碳的距离分别拉长至3.202 和 3.632 Å.由于C11中金属中心失去π配位,自由能升高26.5 kcal/mol(相对于C10).随后,C11经过一个七元环状过渡态(TSC6a)还原消除得七元杂环产物(C12),相对于C10,该还原消除的位垒为32.2 kcal/mol,有些偏高.由于在类似的反应中,我们发现前人也报道了偏高的还原消除位垒(高于35.0 kcal/mol)[26-27],可能是由于高斯在计算中高估了Rh与双键间的配位能导致的.C12进一步转变为更稳定的C13,在C13中,七元环生成,相对于能量零点,整个反应的自由能降低28.7 kcal/mol.若由C10直接还原消除得产物,相对于C10,位垒为33.4 kcal/mol(TSC6b).我们对底物1a是否能生成五元螺环产物也进行了探究,计算结果表明,生成五元螺环产物的过渡态(TSC6c)位垒要比生成七元含氧杂环高2.1 kcal/mol,解释了为什么eq1 反应不生成五元螺环产物.之后Rh(I)被Cu(OAc)2氧化为Rh(Ⅲ)重新回到催化循环中,Cu(OAc)被O2及反应中生成的两分子HOAc(分别来自O—H键去质子化和C—H键活化)氧化再生为Cu(OAc)2[28].Seoane及其合作者[16]对该反应进行了氘代试验,结果表明在决速步中有H原子的参与,认为C—H键活化是决速步.根据我们的计算结果,因为C—H活化的位垒比还原消除位垒低14.3 kcal/mol,不可能是决速步,因此我们推测整个催化循环的决速步在催化剂再生.催化剂的氧化再生需要Cu(OAc)2,Cu(OAc)2的再生需要用到HOAc,而HOAc中的H源于反应物C—H键活化,从而也可解释氘代试验.作者在文章中也提到,该体系的反应速率与Cu(OAc)2·H2O量的多少密切相关[16],当Cu(OAc)2·H2O为0.5当量时,反应转化率在1h内可达到97%,当Cu(OAc)2·H2O为0.1当量时,经过16h,反应转化率为87%,速率为前者的1/16,这也表明催化剂再生可能是整个催化循环的决速步.

|
Download:
|
| 图 3 催化[5+2]环加成反应中炔烃迁移插入和还原消除的反应自由能曲线 Fig. 3 Free energy profiles for the migratory insertion of alkyne and the reductive elimination sequences of the Cp*Rh(OAc)2-catalyzed hetero-[5+2] cycloaddition | |
{kind=link}
如图 4所示,在底物1a中有3类氢(Ha、Hb、Hc)可被活化,分别对其活化可以生成五元(A),六元(B)及七元(C)含氧杂环产物.我们对这3条可能的反应途径分别进行计算,结果如图 5所示.由图可以看出,生成A,B产物的反应机理与生成C的相似,均是通过O—H键断裂、C—H键活化、炔烃迁移插入及还原消除4步进行.由于O—H键断裂的位垒极低,炔烃的迁移插入为不可逆过程,因此反应的化学选择性主要由C—H键活化及炔烃迁移插入决定.Path A、Path B、Path C3条反应途径中C—H键活化过渡态自由能依次降低,相对于能量零点,分别是28.0、23.8、17.9 kcal/mol.通过对比3条途径中的过渡态TSA2/TSB2/TSC4a,可看出,虽然在这3条途径中C—H键活化均是通过协同的六元环状过渡态实现,但在TSA2中存在由Rh—C5—C4—O构成的四元环,在TSB2中存在由Rh—C2—C3—C4—O构成的五元环,在TSC4a中存在由Rh—C1—C2—C3—C4—O构成的六元环.由于环张力依次减小,因此,TSA2、TSB2、TSC4a的自由能依次降低.同样,Path A、 Path B、 Path C3条反应途径中的反应位垒依次降低,相对于能量零点,分别是31.0、23.6、20.1 kcal/mol.对比3条途径中炔烃迁移插入的过渡态TSA3/TSB3/TSC5a,炔烃均是通过四元环过渡态迁移插入至Rh—C间,但在3个过渡态中同样依次存在四元、五元及六元环,由于这3类环的稳定性依次增强,因此TSA4、TSB3、TSC5a的自由能依次降低.综上所述,由于环张力的影响,在3条途径中,生成苯并呋喃类产物(A)的反应能垒最高,生成苯并氧杂环庚三烯及其衍生物(C)路径是最有利的.这与实验相符,因作者在文章中指出,以Rh(Ⅲ)为催化剂催化此类反应,迄今为止还没有生成苯并呋喃类产物的报道,而在这个反应体系中,仅检测到产物C,而没有检测到A和B.

|
Download:
|
| 图 4 Cp*Rh(OAc)2催化底物1a与炔烃的环加成反应 Fig. 4 Different annulation options for 1a using Cp*Rh(OAc)2 catalyst | |
{kind=link}
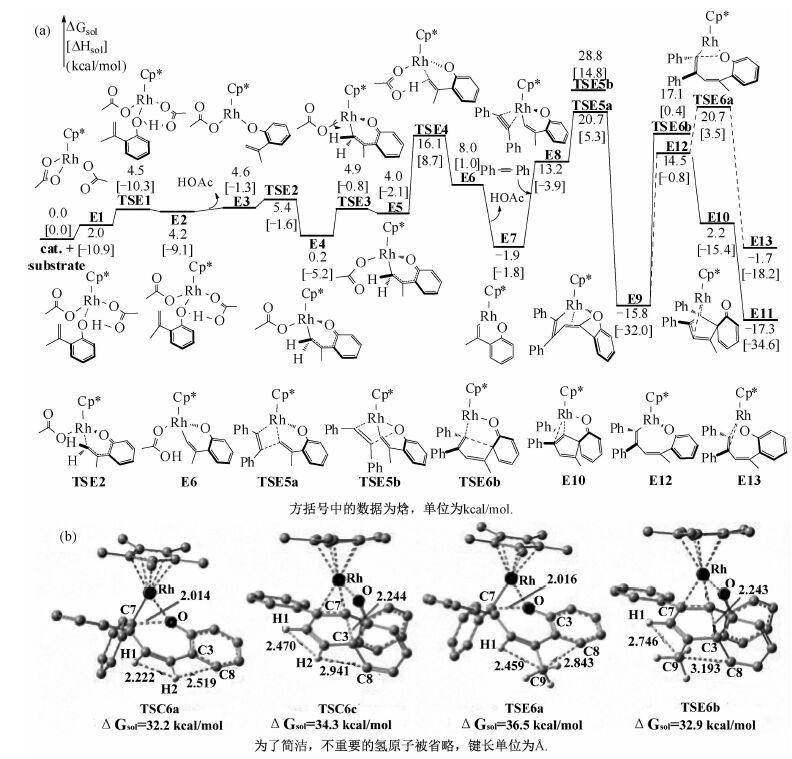
图 6(a)描述底物2a(2-异烯丙基苯酚)与二苯乙炔反应生成五元螺环产物的反应机理.与生成七元含氧杂环相似,该反应同样分为O—H键断裂、C—H键活化、炔烃迁移插入及还原消除.前3步与前一[5+2]反应类似且容易发生,相对于能量零点,3步反应过渡态的自由能分别为4.5 kcal/mol(TSE1)、16.1 kcal/mol(TSE4)、20.7 kcal/mol(TSE5a).炔烃迁移插入后生成E9,经五元环状过渡态(TSE6b)生成五元螺环产物E10,位垒为32.9 kcal/mol(相对于E9).在TSE6b中,原苯酚苯环的芳香性被破坏,Rh—O键断裂,与苯基相连的碳原子与原酚羟基邻位碳成键.之后E10异构化为更稳定的E11(ΔG=-17.3 kcal/mol).计算表明,由E9生成七元含氧杂环产物(E13)的位垒为36.5 kcal/mol(TSE6a),比生成五元螺环高3.6 kcal/mol.之后氧化剂的再生与前一[5+2]反应相同.Seoane及其合作者[16]同样对该反应进行了氘代试验,结果表明在决速步中同样有H原子的参与.同2.1所述原因,我们认为催化剂的再生同样为该反应的决速步.

|
Download:
|
| 图 5 Cp*Rh(OAc)2催化底物1a与二苯乙炔环加成反应中,对不同C—H键活化所得的3条自由能曲线 Fig. 5 Free energy profiles for the activation of the three different C—H bonds of the Cp*Rh(OAc)2-catalyzed cycloaddition of 1a with 1,2-diphenylethyne | |
{kind=link}
底物1a(2-羟基苯乙烯)与2a(2-异烯丙基苯酚)相似,差别仅在于2a比1a在烯基部分多了1个甲基,但是2个反应的产物完全不同.对两例反应的反应机理的对比研究表明,由还原消除最终决定产物的选择性.因此我们对两例反应中决定产物选择性的4个还原消除过渡态(TSC6a/TSC6c,TSE6a/TSE6b)分别进行对比研究.通过对比TSC6a/TSC6c的几何结构(图 6(b)),我们发现在TSC6a中,CC上H1与H2间的距离为2.222 Å,H2与相邻苯环上C8间的距离为2.519 Å,分别略小于氢氢及碳氢间范德华半径(2.40 和2.70 Å),均存在立体排斥.在TSC6c中,H1与H2,H2与C8间的距离分别为2.470 和2.941 Å,不存在排斥力.但是在TSC6c中,与CC相邻的苯环的芳香性被破坏,因此,尽管TSC6c结构中的空间位阻比TSC6a小,但TSC6c中的苯环芳香性被破坏,其对能量的影响大于位阻因素,因此能量比TSC6a高,反应趋向于生成七元杂环产物.对比TSE6a/TSE6b的几何结构,可以清楚地看到,在TSE6a中,CC上的甲基与相邻苯环间有强排斥力,C9与C8间的距离仅为2.843 Å,远小于碳碳间的范德华半径(3.40 Å).同时,C9与相邻的H1间的距离为2.459 Å,也存在排斥力.而在TSE6b中,虽然其与双键相邻的苯环芳香性被破坏,但由于苯环与双键转离同一平面,C9与C8间的距离增加到3.193 Å,排斥力明显减弱.同时,C9与H1间的距离拉长为2.746 Å.因此,即使TSE6a的芳香性保留,但结构中的空间位阻过大,为决定因素,TSE6a能量比TSE6b高,反应更易于生成五元螺环产物.以上对比研究表明,当底物烯基部分无取代基时,在还原消除过渡态中,芳香性是否被破坏为决定因素,反应趋向于进行[5+2]环加成,生成七元含氧杂环产物(因其原底物的芳香性可保留).若底物烯基部分有取代基,则在还原消除过渡态中,取代基的影响使位阻因素起决定作用,反应趋于进行[3+2]环加成,生成五元螺环产物(因其取代基与相邻苯环间排斥力小).

|
Download:
|
| 图 6 (a)Cp*Rh(OAc)2催化[3+2]环加成反应中O—H键的断裂、C—H键活化、炔烃迁移插入和还原消除4步的自由能曲线;(b)2个反应体系中还原消除过渡态的几何结构 Fig. 6 (a)Free energy profiles for the consecutive O—H deprotonation,C—H activation,alkyne insertion,and reductive elimination sequences of the Cp*Rh(OAc)2-catalyzed [3+2] cycloaddition;(b)Geometric structures of TSs for the reductive elimination steps of two kinds of cycloadditions | |
{kind=link}
我们采用密度泛函的方法(B3LYP 与M06连用)对[Cp*RhCl2]2在Cu(OAc)2及氧气存在条件下催化2-羟基苯乙烯与炔烃进行[5+2]环加成生成七元含氧杂环产物,及2-异烯丙基苯酚与炔烃进行[3+2]环加成生成五元螺环产物的反应机理进行了对比研究.研究结果表明,两类反应均是由O—H键断裂、C—H键活化、炔烃迁移插入及还原消除组成.在还原消除时,位阻与芳香性影响的强弱对比决定产物的选择性.当底物为2-羟基苯乙烯时,两过渡态中的空间位阻均不大,而生成七元含氧杂环可使芳香性保留,因此反应趋向于进行[5+2]环加成,当底物为2-异烯基苯酚时,因双键上的甲基使生成七元含氧杂环的过渡态的空间位阻过大,所以反应更易于进行[3+2]环加成.我们以2-羟基苯乙烯与炔烃反应为例,探讨了反应中C—H键活化的选择性,研究结果表明,选择性是由C—H键活化及炔烃迁移插入过渡态中的环张力大小决定的.
| [1] | Lautens M, Klute W, T am, W. Transition metal-mediated cycloaddition reactions[J]. Chem Rev , 1996, 96 (1) :49–92. DOI:10.1021/cr950016l |
| [2] | Rubin M, Rubina M, Gevorgyan V. Transition metal chemistry of cyclopropenes and cyclopropanes[J]. Chem Rev , 2007, 107 (7) :3117–3179. DOI:10.1021/cr050988l |
| [3] | Yeung C S, Dong V M. Catalytic dehydrogenative cross-coupling: forming carbon-carbon bonds by oxidizing two carbon-hydrogen bonds[J]. Chem Rev , 2011, 111 (3) :1215–1292. DOI:10.1021/cr100280d |
| [4] | Wencel D J, Glorius F. C—H bond activation enables the rapid construction and late-stage diversification of functional molecules[J]. Nat Chem , 2013, 5 :369–375. DOI:10.1038/nchem.1607 |
| [5] | Colby D A, Bergman R G, Ellman J A. Rhodium-catalyzed C—C bond formation via heteroatom-directed C—H bond activation[J]. Chem Rev , 2010, 110 (2) :624–655. DOI:10.1021/cr900005n |
| [6] | Guimond N, Gouliaras C, Fagnou K. Rhodium(Ⅲ)-catalyzed isoquinolone synthesis: the N—O bond as a handle for C—N bond formation and catalyst turnover[J]. J Am Chem Soc , 2010, 132 (20) :6908–6909. DOI:10.1021/ja102571b |
| [7] | Ackermann L, Lygin A V, Hofmann N. Ruthenium-catalyzed oxidative annulation by cleavage of C—H/N—H bonds[J]. Angew Chem Int Ed , 2011, 50 (28) :6379–6382. DOI:10.1002/anie.201101943 |
| [8] | Nan J, Zuo Z, Luo L, et al. RuII-catalyzed vinylative dearomatization of naphthols via a C(sp2)—H bond activation approach[J]. J Am Chem Soc , 2013, 135 (46) :17306–17309. DOI:10.1021/ja410060e |
| [9] | Mehta V P, García J A, Greaney M F. Aromatic homologation by non-chelate-assisted RhⅢ-catalyzed C—H functionalization of arenes with alkynes[J]. Angew Chem, Int Ed , 2014, 53 (6) :1529–1533. DOI:10.1002/anie.201309114 |
| [10] | Stuart D R, Bertrand L M, Burgess K M N, et al. Indole synthesis via rhodium catalyzed oxidative coupling of acetanilides and internal alkynes[J]. J Am Chem Soc , 2008, 130 (49) :16474–16475. DOI:10.1021/ja806955s |
| [11] | Rakshit S, Patureau F W, Glorius F. Pyrrole synthesis via allylic sp3 C—H activation of enamines followed by intermolecular coupling with unactivated alkynes[J]. J Am Chem Soc , 2010, 132 (28) :9585–9587. DOI:10.1021/ja104305s |
| [12] | Neely J M, Rovis T. Rh(Ⅲ)-catalyzed regioselective synthesis of pyridines from alkenes and α,β-unsaturated oxime esters[J]. J Am Chem Soc , 2013, 135 (1) :66–69. DOI:10.1021/ja3104389 |
| [13] | Dooley J D, Reddy C S, Lam H W. Catalyst-controlled divergent C—H functionalization of unsymmetrical 2-aryl cyclic 1,3-dicarbonyl compounds with alkynes and alkenes[J]. J Am Chem Soc , 2013, 135 (29) :10829–10836. DOI:10.1021/ja404867k |
| [14] | Cui S, Zhang Y, Wu Q. Rh(Ⅲ)-catalyzed C—H activation/cycloaddition of benzamides and methylenecyclopropanes: divergence in ring formation[J]. Chem Sci , 2013, 4 (9) :3421–3426. DOI:10.1039/c3sc51424b |
| [15] | Zhou M B, Song R J, Wang C Y, et al. Synthesis of azepine derivatives by silver-catalyzed cycloaddition of g-amino ketones with alkynes[J]. Angew Chem Int Ed , 2013, 52 (41) :10805–10808. DOI:10.1002/anie.201304902 |
| [16] | Seoane A, Casanova N, Quiñones N, et al. Straightforward assembly of benzoxepines by means of a rhodium(Ⅲ)-catalyzed C—H functionalization of o-vinylphenols[J]. J Am Chem Soc , 2014, 136 (3) :834–837. DOI:10.1021/ja410538w |
| [17] | Seoane A, Casanova N, Quiñones N, et al. Rhodium(Ⅲ)-catalyzed dearomatizing (3 + 2) annulation of 2-alkenylphenols and alkynes[J]. J Am Chem Soc , 2014, 136 (21) :7607–7610. DOI:10.1021/ja5034952 |
| [18] | Becke A D. Density-functional thermochemistry. Ⅲ. The role of exact exchange[J]. J Chem Phys , 1993, 98 (7) :5648–5652. DOI:10.1063/1.464913 |
| [19] | Roy L E, Hay P J, Martin R L. Revised basis sets for the LANL effective core potentials[J]. J Chem Theory Comput , 2008, 4 (7) :1029–1031. DOI:10.1021/ct8000409 |
| [20] | Dang Y F, Qu S L, Wang Z X, et al. Mechanism and origins of Z selectivity of the catalytic hydroalkoxylation of alkynes via rhodium vinylidene complexes to produce enol ethers[J]. Organometallics , 2013, 32 (9) :2804–0813. DOI:10.1021/om400227u |
| [21] | Dang Y F, Q u, S L, Wang Z X, et al. A computational mechanistic study of an unprecedented heck-type relay reaction:insight into the origins of regio- and enantioselect-ivities[J]. J Am Chem Soc , 2014, 136 (3) :986–998. DOI:10.1021/ja410118m |
| [22] | Qu S L, Dang Y F, Wen M W, et al. Mechanism of the methyltrioxorhenium-catalyzed deoxydehydration of polyols:a new pathway revealed[J]. Chem Eur J , 2013, 19 (12) :3827–3832. DOI:10.1002/chem.201204001 |
| [23] | Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09 . Gaussian, Inc.: Wallingford, CT, 2009. http://www.oalib.com/references/17164148 |
| [24] | Xu L, Zhu Q, Huang G, et al. Computational elucidation of the internal oxidant-controlled reaction pathways in Rh(Ⅲ)-catalyzed aromatic C—H functionalization[J]. J Org Chem , 2011, 76 (9) :3523–3526. DOI:10.1021/jo1025546 |
| [25] | Lapointe D, Fagnou K. Overview of the mechanistic work on the concerted metallation-deprotonation pathway[J]. Chem Lett , 2010, 39 (11) :1118–1126. DOI:10.1246/cl.2010.1118 |
| [26] | Liu L, Wu Y, Wang T, et al. Mechanism, reactivity, and selectivity in Rh(Ⅲ)-catalyzed phosphoryl-directed oxidative C—H activation/cyclization:a DFT study[J]. J Org Chem , 2014, 79 (11) :5074–5081. DOI:10.1021/jo500616g |
| [27] | Quinones N, Seoane A, arcia-Fandino R, et al. Rhodium(Ⅲ)-catalyzed intramolecular annulations involving amide-directed C—H activations: synthetic scope and mechanistic studies[J]. Chem Sci , 2013, 4 (7) :2874–2879. DOI:10.1039/c3sc51078f |
| [28] | Hosokawa T, Murahashi S. New aspects of oxypalladation of alkenes[J]. Acc Chem Res , 1990, 23 (2) :49–54. DOI:10.1021/ar00170a006 |