 2022, Vol. 57
2022, Vol. 57



脊髓性肌萎缩(spinal muscular atrophy, SMA) 是一种严重的神经肌肉疾病, 其特征是脊髓运动神经元变性、骨骼肌萎缩和全身无力[1]。作为一种常见的常染色体隐性神经退行性疾病, 其在新生儿中的发病率估计在1∶6 000至1∶10 000[1, 2]。SMA患者的临床表现不尽相同, 根据发病年龄和达到的最大运动指标将SMA分为3种亚型, 从最严重的Ⅰ型到最轻微的Ⅲ型[3]。其中, 最常见的是Ⅰ型(也被称为Werdnig-Hoffman病), 约占总SMA患者的50%, 大多数患者在婴儿期出现肌无力和早期运动指标的延迟或缺失(例如, 不能在没有帮助的情况下坐着), 在其出生后的前几个月就会出现症状。在没有外部治疗的情况下, 出生后的第二年通常会发生死亡[4, 5]。
SMA的发病机制(图 1) 是由于位于5q染色体13位点上的运动神经元生存基因1 (survival motor neuron gene1, SMN1) 的缺失或突变, 导致其编码的SMN蛋白水平降低, 引起α神经元的缺失和进行性肌肉萎缩[6-8]。SMN蛋白是一种广泛表达的38 kD蛋白, 参与多种细胞功能, 包括pre-mRNA剪接、小核糖核蛋白(small nuclear ribonucleoprotein, snRNP) 生成、转录、应激反应、凋亡、轴突运输和细胞骨架动力学[9]。

|
Figure 1 The disease etiology of SMA[13]. a) Healthy individual. There are two SMN genes in healthy individuals: SMN1 can produces all full length SMN proteins, while SMN2 produces 10% full length SMN proteins and 90% truncated SMN proteins; b) SMA patient. Due to mutations of SMN1, no SMN proteins produced from SMN1, only 10% full length SMN proteins produced from SMN2 |
在人类中存在两种SMN基因: SMN1和SMN2。其中, SMN1产生全长mRNA (full length mRNA, FL mRNA) 转录本, 编码具有完全功能的SMN蛋白; SMN2的基因序列除了其中外显子7第六位的C替换T外, 与SMN1的基因序列相同。因而90%的SMN2基因产生的mRNA转录本编码产生缺乏外显子7 (SMN ∆7) 的蛋白, 此蛋白是无功能的, 并被迅速降解; 只有10%功能性SMN蛋白产生。SMN2拷贝的数量与临床表型呈负相关[10-13]。在SMN1缺失的情况下, 虽然SMN2的存在能够挽救胚胎致死, 但其不足以防止运动神经元的退化[14]。
允许从单个基因表达多个mRNA亚型, 真核细胞的选择性剪接增加基因组的编码潜力。剪接可以单独控制每个基因及其部分, 包括外显子包含、外显子跳跃、内含子保留、可选择性5′SS和3′SS的招募以及外显子的相互排斥[15]。一般来说, 内含子的去除是由剪接体完成的, 剪接体的组装包括U1 snRNP、U2AF (U2 snRNP auxiliary factor) 和U2 snRNP对5′SS、3′SS和分支点序列的识别等因素。若存在外显子编码序列的突变, 这个过程中的几个步骤可能会失败[16]。
从SMN1到SMN2, pre-mRNA外显子7第六位发生C-to-T的替换, 这使得SMN2 pre-mRNA在剪接过程中外显子7发生跳跃[17]。这种C-to-T转变破坏了外显子剪接增强子(exonic splicing enhancer, ESE), 同时产生外显子剪接沉默子(exonic splicing silencer, ESS), 其与一种已知的抑制蛋白核不均一核糖核蛋白(heterogeneous nuclear ribonucleoprotein, hnRNP) 结合以抑制外显子7剪接[18]。
在SMA被首次描述后的几十年, 以呼吸机、营养和外科干预的形式提供护理支持, 提高了患者的寿命和舒适度, 然而预后仍然很差[4]。
近年来, 发现了许多可以诱导SMA患者来源的成纤维细胞增加SMN蛋白生成的化合物, 如: aclarubicin、butyrate、4-phenylbutyrate、valproic acid、hydroxyurea、indoprofen、interferons-β and -γ、forskolin、ortho-vanadate、cantharidin、tautomycin、aminoglycosides、resveratrol、suberoylanilide hydroxamic acid and M344。由于这些化合物需要高药物浓度才能增加SMN蛋白表达, 在体内代谢迅速或具有毒性, 许多化合物也无法透过血脑屏障, 导致其中许多化合物不能成为良好的治疗药物[19]。
随着对SMA遗传原因的深入了解, 研究人员开发了许多新的治疗方案用于治疗这种毁灭性的疾病。Onasemnogene abeparvovec (ZolgenmaTM) 是一种单剂量、静脉注射的基因疗法药物, 其利用非复制、非整合、重组腺相关病毒血清型9 (adeno-associated virus serotype 9, AAV9) 衣壳来传递稳定且功能完全的人SMN转基因, 替换突变或缺失的SMN1基因, 以增加全长SMN蛋白的表达, 改善神经元和肌肉功能, 全疗程仅需一次注射[20-22]。2019年5月, 该药物获得美国FDA批准, 用于治疗诊断为SMA的患儿[23]。2019年其一次注射治疗的费用为212.5万美元, 成为有报道以来世界上最昂贵的药物[24]。
在基因治疗发展的同时, 反义寡核苷酸和小分子药物疗法的发展也取得了重大突破性进展。如增强启动子的活性或SMN2 mRNA转录本中外显子7的包含, 在体内基因调控的不同水平下增加SMN2表达, 进而提高全长SMN蛋白的水平, 改善疾病的症状。下面简要介绍几种具有代表性的作用于SMN2的反义寡核苷酸药物和小分子药物。
1 作用于SMN2的药物作用于SMN2的药物主要分为两类: 小分子化学药物和反义寡核苷酸药物。反义寡核苷酸药物需要通过注射给药, 而小分子药物则可口服给药, 具有口服给药的固有优势以及广泛的外周和中枢神经系统(central nervous system, CNS) 分布[25]。下面将按照治疗药物所处研发阶段加以总结介绍。
1.1 已上市药物 1.1.1 诺西那生钠(nusinersen)反义寡核苷酸(antisense oligonucleotides, ASOs) 是一种与特定pre-mRNA具有互补序列的RNA分子, 并可与其碱基互补配对结合, 干扰正常蛋白质-RNA或RNA-RNA相互作用而调节剪接[26]。
Nusinersen (SpinzaraTM, 即ASO-10-27 5′-TCACT TTCATAATGCTGG-3′) 是由Ionis Pharmaceuticals和百健(Biogen) 共同开发的一种2′-O-(2-甲氧基乙基) (MOE) 磷酸修饰的反义寡核苷酸, 靶向内含子7位点的单链DNA, 用于治疗SMN蛋白水平不足引起的SMA[27]。2016年12月, 美国FDA正式批准其用于所有类型SMA的治疗, 成为获批的首个用于该适应症治疗的药物[28]。2019年4月, 其在中国上市, 成为中国首个治疗SMA的药物。该药需由具有腰椎穿刺经验的医疗专业人员或其指导下进行鞘内给药[28]。
在SMA发病机制的调控网络中, 位于SMN2外显子7下游5′剪接位点的内含子7剪接沉默子(intronic splicing silencer, ISS) 是调节外显子7可选择性剪接的重要组成部分。其中, ISS-N1的功能是募集抑制外显子7包含的正常剪接抑制因子hnRNP A1和A2[29]。Nusinersen通过序列特异性的碱基互补配对, 与SMN2 pre-mRNA上ISS-N1位点结合, 阻断了hnRNA蛋白的募集, 使SMN2外显子7在pre-mRNA剪接中被包含, 增加了全长SMN蛋白的量[30]。Nusinersen被誉为“非常有效”, 它的两名发明者获得了300万美元的“突破奖”[24]。如图 2[31]所示。

|
Figure 2 The mechanism of action of nusinersen[31]. Nusinersen binds to the intron splicing silencer N1 (ISS-N1) site on the SMN2 pre-mRNA. By displacing hRNP A1/2 from the ISS-N1 site, nusinersen promotes the inclusion of exon7. ASO: Antisense oligonucleotide |
Nusinersen注射液在美国的单支价格为12.5万美元, 我国的价格约合70万人民币/支。在前两个月完成4次剂量注射后, 终生需每四个月注射一次维持治疗, 其治疗费用极其昂贵。2021年12月, 国家医保局公布了2021年国家医保药品目录, nusinersen新增进入目录, SMA患者的用药费用将显著降低[32]。
1.1.2 利司扑兰(risdiplam)Risdiplam (EvrysdiTM) 是由罗氏(Roche)、PTC治疗公司以及SMA基金会共同开发的口服RNA剪接修饰剂, 口服使用该药能促进SMN2产生全长和功能性的蛋白。
研究者们使用含有SMN2微小基因的HEK293H人胚胎肾细胞系, 通过高通量筛选(high-throughput screening, HTS) 的方法从含有20多万种化合物的库中, 筛选出了低效力的香豆素衍生物1 (图 3)。对其进一步优化后, 得到两个口服可利用的SMN化合物系列: 异香豆素衍生物SMN-C1、香豆素衍生物SMN-C2, 可以特异性地对SMN2 pre-mRNA进行修饰[9]。然而, 由于其在血浆和水缓冲液中存在光毒性、遗传毒性和化学不稳定性, 研究因此停止[33]。

|
Figure 3 The structures of risdiplam and its related compounds |
随后, 研究者们以吡啶并嘧啶酮为母核进行优化, 其中代表性的化合物是SMN-C3和RG7800[33]。由于RG7800表现出良好的药代动力学性质和体内药效, 其成为第一个进入人体临床试验的小分子SMN2剪接修饰剂[33]。在健康人体进行的Ⅰ期临床试验中, 研究者对RG7800的安全性、耐受性、药代动力学(pharmacokinetics, PK) 进行了研究, 该化合物在所有试验剂量下均安全且耐受良好[34]。然而, 在与临床研究平行开展的长期慢性毒性研究中, 发现长期口服给药的食蟹猴出现视网膜毒性, 其临床研究被迫停止[35]。
研究者们对此类化合物进行了更加深入的优化研究, risdiplam因为具有非常高的效力、显著降低的碱性、无光毒性风险且代谢物少的特点而被选择用于进一步的研究[34]。2020年8月, risdiplam获得美国FDA批准, 用于2个月及以上年龄患者的脊髓性肌萎缩治疗[36], 也是第一个被批准作为SMA剪接修饰剂的药物[37]。
如图 3所示, 此类化合物的构效关系(structure-activity relationship, SAR) 可简要归纳如下: 相比香豆素和异香豆素母核, 吡啶并嘧啶酮母核在pH 1到pH 12的人血浆和水缓冲液中化学稳定, 因此光毒性显著降低; Ames实验的结果取决于右侧片段(R1), 在吡啶并嘧啶酮母核的基础上, 吡唑并吡嗪或取代的咪唑并吡啶部分作为右侧片段, 体外效力良好, Ames实验阴性, 右侧片段以咪唑并哒嗪为最好; 左侧片段(R2) 的特定位置[SMN-C3左侧片段(1-乙基哌啶-4-基) N原子位置] 引入碱性N原子效力好, risdiplam相比RG7800, 口服后不会产生N-脱烷基代谢物, 不会诱导FoxM1 (forkhead box protein M1) 介导的潜在的外周不良反应。
如图 4所示, 在pre-mRNA剪接的过程中, U1snRNP与位于外显子-内含子边界高度保守的5′剪接位点(5′SS) 的杂交是非常关键的[38]。在SMN2 pre-mRNA的剪接过程中, 5′SS的特殊替换使其与U1snRNP的结合减弱, 直接导致剪接跳过[38]。Risdiplam类药物特异性地稳定由5′SS和U1snRNP复合物形成的瞬时双链RNA结构, 从而将SMN2外显子7的弱5′SS转化为强5′SS, 有利于剪接的进行。同时, 此类化合物还可以选择性地与SMN2外显子7中的外显子剪接增强子ESE2结合, 以增强靶向SMN2的特异性[16]。Risdiplam与SMN2外显子7的5′SS以及ESE2的结合, 使其对SMN2靶标具有特异性[38]。在来源于患者的成纤维细胞以及由诱导多能干细胞(induced pluripotent stem cells, iPSCs) 产生的运动神经元细胞中, risdiplam促进了外显子7的包含[30], 产生全长的FL mRNA, 使全长SMN蛋白增加[34]。

|
Figure 4 The mechanism of action of risdiplam[38]. A: Binding to the transient double-strand RNA structure formed by 5′SS and U1 snRNA (the RNA component of the U1 snRNP), risdiplam converts the weak 5′SS into a stronger one; B: Risdiplam binds to 5′SS as well as to ESE2 of SMN2 exon 7. SM: Risdiplam |
自2020年8月在美国FDA正式获批后, risdiplam在SMA治疗的药物市场份额一直在增长, 第三季度的销售额为890万美元[37]。
相比于nusinersen需要通过鞘内给药至中枢神经系统, risdiplam通过口服方式给药, 可遍布中枢神经系统和外周组织[39]。这对治疗疾病来说是有利的, 因为有证据表明外周中SMN蛋白水平对疾病状态有很大的影响[40, 41]。
1.2 处于临床研究阶段的候选药物 1.2.1 BranaplamBranaplam是诺华公司研发中的SMN2剪接修饰剂。通过使用表达SMN2微小基因报告基团的NSC34运动神经元细胞系进行高通量筛选, 从诺华化合物库(约1.1×106个) 中鉴定出以哒嗪母核作为核心功能的结构骨架, 对其结构进行优化, 得到NVS-SM1 (branaplam)、NVS-SM2这两个具有良好活性的化合物(图 5)。图 6简要总结描述了此类化合物的构效关系。通过ELISA评价SMN ∆7小鼠成肌细胞中SMN蛋白水平, branaplam和NVS-SM2的EC50分别为0.02和0.6 μmol·L-1 [42]。

|
Figure 5 The structures of NVS-SM1 and NVS-SM2 |

|
Figure 6 SAR of pyridazine series |
为了探索两个化合物是否具有体内活性, 研究者们使用C/+SMA小鼠模型进行评估。口服给药后, 两种化合物在脑和脊髓均产生了剂量依赖性的SMN2-FL转录本以及SMN蛋白的增加。相对而言, NVS-SM1在较低的剂量和暴露下表现出了有效性。同时, 使用NVS-SM1在严重的SMA临床前模型(SMN ∆7) 中证明了持续的体内有效性[42]。基于临床前研究数据, 选择了NVS-SM1进入临床研究, 目前Ⅱ期临床试验已经完成[35]。
NVS-SM1具有改变剪接活性的作用, 它可以通过稳定U1-snRNP复合物与SMN2 pre-mRNA之间的相互作用, 促进外显子7的包含, 产生具有功能的全长SMN蛋白[42]。
Ⅱ期临床研究(NCT02268552) 是一项开放标签、多部分、首次在人体内对Ⅰ型SMA婴儿口服branaplam的研究: 目的是评估13周后的安全性、耐受性、药代动力学(pharmacokinetics, PK)、药效学(pharmacodynamics, PD) 和疗效, 评估口服branaplam的最大耐受量(maximal tolerable dose, MTD), 确定Ⅰ型SMA患者长期使用的安全剂量以及提供持久疗效的最佳剂量方案[43]。然而, 一项长期动物毒性研究结果发现, 每天按规定剂量给药, 为期1年, 观察到外周神经、脊髓、睾丸和肾脏血管的意外损伤, Ⅱ期临床试验因此暂停[35]。
1.2.2 SMN-C3前文所述, 对通过HTS获得的先导化合物香豆素衍生物1 (图 3) 进行优化得到3个类别的化合物, SMN-C3是其中一个类别的代表性化合物[9, 33]。
该化合物具有纳摩尔水平的效价(EC1.5x = 84 nmol·L-1), 增加了Ⅰ型SMA患者的成纤维细胞FL SMN2 mRNA水平, 同时降低了∆7 mRNA的水平, 从而SMN蛋白表达增加[9]。在患者来源的诱导多能干细胞产生的Islet-1阳性运动神经元细胞和Islet-1阴性运动神经元细胞中, 使用SMN-C3也可增加Ⅰ型和Ⅱ型SMA患者细胞中的SMN蛋白水平[9]。在严重SMA小鼠模型中, 出生后早期使用SMN2剪接修饰剂可防止出现肌萎缩, 同时还能延长寿命、增加体重, 预防成年后的运动功能障碍和神经肌肉缺陷[9]。
当U1snRNA结合时, SMN-C类化合物结合到SMN2外显子7上5′SS和ESE2两个位点: 与5′SS结合促进了外显子7在剪接过程中的包含, 与ESE2结合导致hnERP (heterogeneous nuclear ribonucleoprotein) G错位并允许U1snRNP复合物与ESE2结合。两个结合过程使得SMN-C类化合物显示对SMN2及STRN3 pre-mRNA的高选择性[16]。
1.2.3 RG3039在HTS实验中, 使用基于细胞的SMN2转录小分子激动剂报告基因分析, 喹唑啉类化合物被鉴定出来作为运动神经元类细胞系的SMN2基因转录激活剂[44]。基于配体的设计策略开发了一系列C5取代的2, 4-二氨基喹唑啉类化合物, 其中一些化合物在SMA患者来源的成纤维细胞系表现SMN mRNA和全长SMN蛋白水平的提高以及细胞核内Cajal体(cajal body) 和Gem (gemini of coiled body) 数量的增加[45]。用严重SMA小鼠模型delta-7小鼠测试了代表性化合物D156844 (图 7), 口服该化合物可延长动物平均寿命, 并改善运动表型[19, 46]。在进一步的药物化学优化中, D157495 (RG3039, PF-06687859) 与其他化合物相比没有脱靶风险而被选为临床候选药物[27, 47]。

|
Figure 7 The structures of D156844 and RG3039 |
Gem缺失是来源于SMA患者的成纤维细胞特征, 其数量与疾病严重程度呈负相关[47]。使用放射性标记的C5取代的喹唑啉进行蛋白微阵列扫描, 此类化合物的分子靶点被确定为清道夫脱帽酶(decapping scavenger enzyme, DcpS)。DcpS是一种结合和水解m7GpppN mRNA帽结构的核穿梭蛋白, 是RNA代谢的调节剂。此类化合物与DcpS结合使酶处于一种开放的、无法催化的构象中, 有效抑制DcpS脱帽活性[48]。
到目前为止, 包含32位志愿者的双盲Ⅰ期临床试验已经完成, RG3039在所有试验剂量下均可被耐受, 没有不良反应事件[49]。
1.3 处于临床前研究阶段的药物候选物 1.3.1 苯基噻二唑类化合物前文提到, branaplam是诺华公司研发的选择性SMN2剪接修饰剂, 目前处于Ⅱ期临床研究阶段。如图 8所示, 其结构哒嗪-苯基联芳基体系的近共平面构象是由哒嗪与苯酚形成的分子内氢键所强制形成的, 这对维持其活性是必须的。构效关系显示, 哒嗪可被1, 3, 4-噻二唑取代, 这为联芳基通过分子内1, 5-硫-氧(S···O) 或1, 5-硫-卤(S···X) 非共价相互作用提供了构象约束的其他机会[41]。代表化合物及其活性数据如表 1所示。

|
Figure 8 Conformational constraints. A: 2-Hydroxyphenyl pyridazine; B: 2-Hydroxyphenyl thiadiazole; C: 2-Methoxyphenyl thiadiazole |
| Table 1 SAR of phenyl thiadiazoles |
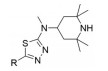



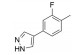

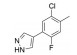

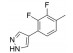
G-2-1和G-2-2的PK数据表明, 氟或氯取代加上4-(4-吡唑基)苯基取代导致体内清除显著减少, 相对于甲氧基和羟基取代类似物G-1-1、G-1-2和G-1-3具有更高的血浆暴露。G-2-5包含2-氟苯基噻二唑基, 在小鼠SMA模型中显示全长SMN蛋白的产量增加了50%以上[41]。
1.3.2 PK4C9在调控外显子7包含的顺式调控元件中, 位于SMN2转录本上外显子7与内含子7连接处的TSL2 (terminal stem-loop 2) 发挥着关键作用[50, 51]。有人提出, TSL hairpin的3′端部分封闭了E7的5′剪接位点, 对剪接机制的招募和外显子的包含产生了不利影响[50]。研究人员使用TSL2 RNA结构作为靶标, 对SMA进行基于靶标的小分子筛选, 确定出了一类通过稳定TSL2构象, 提高5′剪接位点的可及性来增加SMN2 E7剪接的先导化合物。海洋天然产物homocarbonyltopsentin (PK4C9, 图 9) 是其中最强效的化合物, 其在40 μmol·L-1时E7的平均包含率达72% (与DMSO处理的细胞相比增加了43%), EC50值是25 μmol·L-1。用PK4C9 (40 μmol·L-1, 48 h) 处理Ⅰ型SMA患者的成纤维细胞后, SMN蛋白含量较DMSO处理后增加1.5倍。但是这种效应依赖于TSL2的完整性, 携带TSL2结构突变的微小基因的使用影响了PK4C9的活性[52]。

|
Figure 9 The structure of PK4C9 |
如图 10所示, TSL2的三环构象(triloop conformation) 和五环构象(pentaloop conformation) 共存, 其中三环构象对5′SS和E7剪接的接近能力更好。PK4C9会与pentaloop TSL2结合, 直接促进5′SS的暴露, 并间接引起triloop的转移。PK4C9通过诱导TSL2局部开放以提高E7的5′SS对剪接因子的可及性[52]。

|
Figure 10 The Mechanism of action of PK4C9[52] |
为了鉴定可促进SMN表达的诱导物, 来自印第安纳大学医学院(Indiana University School of Medicine) 等机构的研究者们开发了一种基于细胞的SMN2报告基因实验, 确定了先导化合物LDN-75654 (EC50 = 1.8 μmol·L-1) (图 11)。虽然LDN-75654可促进SMN在SMA患者来源的成纤维细胞中的表达增加, 但由于其代谢稳定性差和溶解性差导致其在体内表现不佳。对LDN-75654类化合物进行结构优化, 得到化合物4m (EC50 = 0.36 μmol·L-1) 和LDN-2014 (EC50 = 0.29 μmol·L-1) (图 11)。然而, 最有希望的4m具有不利的PK特性。在生物利用度驱动的异噁唑系列优化中, 酰胺连接被证明与较差的血浆稳定性有关, 但可以通过改变杂环和逆转酰胺键而得到改善, LDN-2014在注射后表现出脑暴露量改善[53]。

|
Figure 11 The structures of LDN-75654, 4m and LDN-2014 |
虽然LDN-2014改变蛋白水平的机制尚不明确, 但其确实改变了SMN蛋白的水平。LDN-2014在重度SMN ∆7和中度SMN2B/-小鼠两种重要的SMA动物模型中具有生物活性[54]。
1.3.4 3, 4-二氢-4-苯基-2(1H)-喹唑啉衍生物不同于之前的SMN2剪接修饰剂, 布列根和妇女医院(Brigham and Women's Hospital) 与哈佛大学医学院(Harvard Medical School) 等单位共同研发了一类3, 4-二氢-4-苯基-2(1H)-喹唑啉衍生物(图 12), 其代表为I-1 (EC50 = 4.1 μmol·L-1), 通过全新的作用机制—在转录水平上调控SMN2的表达, 作为一种SMN2表达增强剂发挥作用[55]。

|
Figure 12 The structure of I-1 |
该化合物不仅在小鼠的微粒体试验中表现出了良好的水溶性和稳定性, 而且在一项针对SMN ∆7小鼠的疗效研究中, 每天服用该化合物, 小鼠的生存率延长了两倍[55]。基于上述研究结果, 该化合物具有良好的前景去进行进一步的研究。
1.3.5 反义寡核苷酸类药物候选物除上述小分子化学药物外, 目前还有许多处于临床前研究阶段的反义寡核苷酸药物候选物, 如表 2所示。
| Table 2 ASOs in preclinical studies |
以nusinersen为代表的当前一代反义寡核苷酸由于无法透过血脑屏障, 需要采用多次鞘内注射的方式给药。虽然鞘内注射是一种安全且简单的方法, 但仍存在头痛、背痛以及短暂或持续的脑脊液渗漏等不良反应[56]。Hammond等[57]报道了Pip6a-morpholino phosphorodiamidate oligomer (PMO), 即主导传递肽Pip6a与20-mer PMO序列结合, 靶向SMN2外显子7的ISS-N1。在静脉注射给药后, 严重SMA小鼠的中枢神经系统和外周组织高效表达SMN蛋白, 对于治疗SMA有极大的潜力。
E1 (Mov11)、PMO-25和HsMN2Ex7D (-10, -29) 的化学组成均为morpholino修饰的寡核苷酸, 靶向SMN2内含子7中内含子抑制因子E1 (element 1) 或ISS-N1, 通过调节SMN2剪接促进外显子7的包含以增加全长SMN蛋白的量, 进而改善SMA症状, 通过注射给药后, 显著延长了严重SMA小鼠的生存期[58-60]。
2 总结与展望作为一种严重的遗传性神经肌肉罕见病, 脊髓性肌萎缩越来越受到社会各界的关注。自发现SMA是由患者的SMN1基因突变或缺失引起的, 并且可以从SMN2基因中增加SMN蛋白的产生以改善疾病症状以来, 在基因治疗、小分子化学药物和反义寡核苷酸药物治疗等方面取得了重大突破。
目前, 关于SMA的治疗药物研究热点主要集中于以下几个方面: ①以nusinersen为代表的反义寡核苷酸药物和以risdiplam为代表的小分子化学药物, 作用于SMN2基因, 调节SMN2的基因转录和剪接修饰过程, 以促进SMN2基因中外显子7的包含来产生完整的全长SMN蛋白。②基因治疗: 以onasemnogene abeparvovec为代表的由腺相关病毒介导的SMN1基因替代疗法, 以弥补SMN1缺失的缺陷。③其他神经保护药物和以肌肉为作用靶点的药物。
随着体外筛选模型的建立, 通过使用高通量筛选技术, 以risdiplam为代表的小分子化学药物成为治疗SMA药物的主要研究方向之一。对各类小分子化合物的作用机制研究, 使得研究者对SMN2基因转录和剪接修饰的影响因素的认识更加深入, 同时, 对SMN2基因转录和剪接修饰机制的进一步阐明, 有助于产生新的筛选策略用于治疗药物的发现。
本文简要介绍了多个作用于SMN2基因的小分子化学药物和反义寡核苷酸药物。已上市的risdiplam和nusinersen由于治疗价格高昂, 广受社会关注, 也使得疗效确切、药物可及性高的新药研究显示出了更高的紧迫性。目前, 多个反义寡核苷酸药物和小分子化学药已处于临床(前) 研究阶段, 进一步的研究正在开展。随着对SMA发病机制的不断持续深入研究, 相信未来一定会有更多新的治疗药物和治疗方式出现, 弥补目前SMA治疗手段的不足, 造福更多的SMA患者。
作者贡献: 贺志昊负责文章的资料收集与撰写; 张翔负责文章的选题与修改。
利益冲突: 作者声明没有任何利益冲突。
| [1] |
Lunn MR, Wang CH. Spinal muscular atrophy[J]. The Lancet, 2008, 371: 2120-2133. DOI:10.1016/S0140-6736(08)60921-6 |
| [2] |
Schorling DC, Pechmann A, Kirschner J. Advances in treatment of spinal muscular atrophy-new phenotypes, new challenges, new implications for care[J]. J Neuromuscul Dis, 2020, 7: 1-13. DOI:10.3233/JND-190424 |
| [3] |
Ramdas S, Servais L. New treatments in spinal muscular atrophy: an overview of currently available data[J]. Expert Opin Pharmacother, 2020, 21: 307-315. DOI:10.1080/14656566.2019.1704732 |
| [4] |
Nance JR. Spinal muscular atrophy[J]. Continuum (Minneap Minn), 2020, 26: 1348-1368. |
| [5] |
Faravelli I, Nizzardo M, Comi GP, et al. Spinal muscular atrophy-recent therapeutic advances for an old challenge[J]. Nat Rev Neurol, 2015, 11: 351-359. DOI:10.1038/nrneurol.2015.77 |
| [6] |
Fallini C, Bassell GJ, Rossoll W. Spinal muscular atrophy: the role of SMN in axonal mRNA regulation[J]. Brain Res, 2012, 1462: 81-92. DOI:10.1016/j.brainres.2012.01.044 |
| [7] |
Schrank B, Gotz R, Gunnersen JM, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos[J]. Proc Natl Acad Sci U S A, 1997, 94: 9920-9925. DOI:10.1073/pnas.94.18.9920 |
| [8] |
Paushkin S, Gubitz AK, Massenet S, et al. The SMN complex, an assemblyosome of ribonucleoproteins[J]. Curr Opin Cell Biol, 2002, 14: 305-312. DOI:10.1016/S0955-0674(02)00332-0 |
| [9] |
Naryshkin NA, Weetall M, Dakka A, et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy[J]. Science, 2014, 345: 688-693. DOI:10.1126/science.1250127 |
| [10] |
Tiziano FD, Bertini E, Messina S, et al. The Hammersmith functional score correlates with the SMN2 copy number: a multicentric study[J]. Neuromuscul Disord, 2007, 17: 400-403. DOI:10.1016/j.nmd.2007.02.006 |
| [11] |
Prior TW, Krainer AR, Hua Y, et al. A positive modifier of spinal muscular atrophy in the SMN2 gene[J]. Am J Hum Genet, 2009, 85: 408-413. DOI:10.1016/j.ajhg.2009.08.002 |
| [12] |
Calucho M, Bernal S, Alias L, et al. Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases[J]. Neuromuscul Disord, 2018, 28: 208-215. DOI:10.1016/j.nmd.2018.01.003 |
| [13] |
Chong LC, Gandhi G, Lee JM, et al. Drug discovery of spinal muscular atrophy (SMA) from the computational perspective: a comprehensive review[J]. Int J Mol Sci, 2021, 22: 8962. DOI:10.3390/ijms22168962 |
| [14] |
Ravi B, Chan-Cortes MH, Sumner CJ. Gene-targeting therapeutics for neurological disease: lessons learned from spinal muscular atrophy[J]. Annu Rev Med, 2021, 72: 1-14. DOI:10.1146/annurev-med-070119-115459 |
| [15] |
Sperling R. The nuts and bolts of the endogenous spliceosome[J]. Wiley Interdiscip Rev RNA, 2017, 8: e1377. |
| [16] |
Sivaramakrishnan M, McCarthy KD, Campagne S, et al. Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers[J]. Nat Commun, 2017, 8: 1476. DOI:10.1038/s41467-017-01559-4 |
| [17] |
Li Q. Nusinersen as a therapeutic agent for spinal muscular atrophy[J]. Yonsei Med J, 2020, 61: 273-283. DOI:10.3349/ymj.2020.61.4.273 |
| [18] |
Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy[J]. Nat Genet, 2003, 34: 460-463. DOI:10.1038/ng1207 |
| [19] |
Butchbach ME, Singh J, Thorsteinsdottir M, et al. Effects of 2, 4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy[J]. Hum Mol Genet, 2010, 19: 454-467. DOI:10.1093/hmg/ddp510 |
| [20] |
Chand D, Mohr F, McMillan H, et al. Hepatotoxicity following administration of onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy[J]. J Hepatol, 2021, 74: 560-566. DOI:10.1016/j.jhep.2020.11.001 |
| [21] |
Connock M, Andronis L, Auguste P, et al. Will the US$5 million onasemnogene abeparvosec treatment for spinal muscular atrophy represent 'value for money' for the NHS? A rapid inquiry into suggestions that it may be cost-effective[J]. Expert Opin Biol Ther, 2020, 20: 823-827. DOI:10.1080/14712598.2020.1772747 |
| [22] |
Liu BN, Xu LC, Han DM, et al. Chemistry, manufacturing and control considerations of rAAV-based gene therapy products[J]. Acta Pharm Sin (药学学报), 2020, 55: 1965-1970. |
| [23] |
Axford J, Sung MJ, Manchester J, et al. Use of intramolecular 1, 5-sulfur-oxygen and 1, 5-sulfur-halogen interactions in the design of N-methyl-5-aryl-N-(2, 2, 6, 6-tetramethylpiperidin-4-yl)-1, 3, 4-thiadiazol-2-amine SMN2 splicing modulators[J]. J Med Chem, 2021, 64: 4744-4761. DOI:10.1021/acs.jmedchem.0c02173 |
| [24] |
Darrow JJ, Sharma M, Shroff M, et al. Efficacy and costs of spinal muscular atrophy drugs[J]. SciTransl Med, 2020, 12: essay9648. |
| [25] |
Woll MG, Qi H, Turpoff A, et al. Discovery and optimization of small molecule splicing modifiers of survival motor neuron 2 as a treatment for spinal muscular atrophy[J]. J Med Chem, 2016, 59: 6070-6085. DOI:10.1021/acs.jmedchem.6b00460 |
| [26] |
Havens MA, Hastings ML. Splice-switching antisense oligonucleotides as therapeutic drugs[J]. Nucleic Acids Res, 2016, 44: 6549-6563. DOI:10.1093/nar/gkw533 |
| [27] |
Van Meerbeke JP, Gibbs RM, Plasterer HL, et al. The DcpS inhibitor RG3039 improves motor function in SMA mice[J]. Hum Mol Genet, 2013, 22: 4074-4083. DOI:10.1093/hmg/ddt257 |
| [28] |
Hoy SM. Nusinersen: first global approval[J]. Drugs, 2017, 77: 473-479. DOI:10.1007/s40265-017-0711-7 |
| [29] |
Singh NK, Singh NN, Androphy EJ, et al. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron[J]. Mol Cell Biol, 2006, 26: 1333-1346. DOI:10.1128/MCB.26.4.1333-1346.2006 |
| [30] |
Chiriboga CA. Nusinersen for the treatment of spinal muscular atrophy[J]. Expert Rev Neurother, 2017, 17: 955-962. DOI:10.1080/14737175.2017.1364159 |
| [31] |
Chiriboga CA, Swoboda KJ, Darras BT, et al. Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy[J]. Neurology, 2016, 86: 890-897. DOI:10.1212/WNL.0000000000002445 |
| [32] |
National Healthcare Security Administration. National Healthcare Security Administration and Ministry of Human Resources and Social Security of the People's Republic of China issued the 2021version of catalog of medicines covered by national medical insurancesystem [EB/OL]. Beijing: National Healthcare Security Administration, 2021 [2021-12-23]. http://www.nhsa.gov.cn/art/2021/12/3/art_14_7430.html.
|
| [33] |
Ratni H, Karp GM, Weetall M, et al. Specific correction of alternative survival motor neuron 2 splicing by small molecules: discovery of a potential novel medicine to treat spinal muscular atrophy[J]. J Med Chem, 2016, 59: 6086-6100. DOI:10.1021/acs.jmedchem.6b00459 |
| [34] |
Ratni H, Ebeling M, Baird J, et al. Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA)[J]. J Med Chem, 2018, 61: 6501-6517. DOI:10.1021/acs.jmedchem.8b00741 |
| [35] |
Ratni H, Mueller L, Ebeling M. Rewriting the (tran)script: application to spinal muscular atrophy[J]. Prog Med Chem, 2019, 58: 119-156. |
| [36] |
Dhillon S. Risdiplam: first approval[J]. Drugs, 2020, 80: 1853-1858. DOI:10.1007/s40265-020-01410-z |
| [37] |
Sheridan C. First small-molecule drug targeting RNA gains momentum[J]. Nat Biotechnol, 2021, 39: 6-8. DOI:10.1038/s41587-020-00788-1 |
| [38] |
Ratni H, Scalco RS, Stephan AH. Risdiplam, the first approved small molecule splicing modifier drug as a blueprint for future transformative medicines[J]. ACS Med Chem Lett, 2021, 12: 874-877. DOI:10.1021/acsmedchemlett.0c00659 |
| [39] |
Calder AN, Androphy EJ, Hodgetts KJ. Small molecules in development for the treatment of spinal muscular atrophy[J]. J Med Chem, 2016, 59: 10067-10083. DOI:10.1021/acs.jmedchem.6b00670 |
| [40] |
Nash LA, Burns JK, Chardon JW, et al. Spinal muscular atrophy: more than a disease of motor neurons?[J]. Curr Mol Med, 2016, 16: 779-792. DOI:10.2174/1566524016666161128113338 |
| [41] |
Hua Y, Sahashi K, Rigo F, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model[J]. Nature, 2011, 478: 123-126. DOI:10.1038/nature10485 |
| [42] |
Palacino J, Swalley SE, Song C, et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice[J]. Nat Chem Biol, 2015, 11: 511-517. DOI:10.1038/nchembio.1837 |
| [43] |
Novartis. An Open Label Study of LMI070 (Branaplam) in Type 1 Spinal Muscular Atrophy (SMA) [EB/OL]. U. S. National library of Medicine: ClinicalTrials. gov, 2021 [2021-10-23].https://clinicaltrials.gov/ct2/show/study/NCT02268552?term=NCT02268552.
|
| [44] |
Jarecki J, Chen X, Bernardino A, et al. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: early leads towards a therapeutic for spinal muscular atrophy[J]. Hum Mol Genet, 2005, 14: 2003-2018. DOI:10.1093/hmg/ddi205 |
| [45] |
Thurmond J, Butchbach ME, Palomo M, et al. Synthesis and biological evaluation of novel 2, 4-diaminoquinazoline derivatives as SMN2 promoter activators for the potential treatment of spinal muscular atrophy[J]. J Med Chem, 2008, 51: 449-469. DOI:10.1021/jm061475p |
| [46] |
Le TT, Pham LT, Butchbach ME, et al. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN[J]. Hum Mol Genet, 2005, 14: 845-857. DOI:10.1093/hmg/ddi078 |
| [47] |
Gogliotti RG, Cardona H, Singh J, et al. The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models[J]. Hum Mol Genet, 2013, 22: 4084-4101. DOI:10.1093/hmg/ddt258 |
| [48] |
Singh J, Salcius M, Liu SW, et al. DcpS as a therapeutic target for spinal muscular atrophy[J]. ACS Chem Biol, 2008, 3: 711-722. DOI:10.1021/cb800120t |
| [49] |
Pharmaprojects. RG3039 [EB/OL]. New York: Informa Business Intelligence Inc, 2016 [2021-12-23].https://citeline.informa.com/drugs/details/66416?qId=e3d19530-0906-440e-b0c1-b13262e9ea7b.
|
| [50] |
Singh NN, Lee BM, Singh RN. Splicing regulation in spinal muscular atrophy by an RNA structure formed by long-distance interactions[J]. Ann N Y Acad Sci, 2015, 1341: 176-187. DOI:10.1111/nyas.12727 |
| [51] |
Disney MD, Yildirim I, Childs-Disney JL. Methods to enable the design of bioactive small molecules targeting RNA[J]. Org Biomol Chem, 2014, 12: 1029-1039. DOI:10.1039/C3OB42023J |
| [52] |
Garcia-Lopez A, Tessaro F, Jonker HRA, et al. Targeting RNA structure in SMN2 reverses spinal muscular atrophy molecular phenotypes[J]. Nat Commun, 2018, 9: 1-12. DOI:10.1038/s41467-017-02088-w |
| [53] |
Rietz A, Li H, Quist KM, et al. Discovery of a small molecule probe that post-translationally stabilizes the survival motor neuron protein for the treatment of spinal muscular atrophy[J]. J Med Chem, 2017, 60: 4594-4610. DOI:10.1021/acs.jmedchem.6b01885 |
| [54] |
Osman EY, Rietz A, Kline RA, et al. Intraperitoneal delivery of a novel drug-like compound improves disease severity in severe and intermediate mouse models of spinal muscular atrophy[J]. Sci Rep, 2019, 9: 1-8. |
| [55] |
Choi S, Calder AN, Miller EH, et al. Optimization of a series of heterocycles as survival motor neuron gene transcription enhancers[J]. Bioorg Med Chem Lett, 2017, 27: 5144-5148. DOI:10.1016/j.bmcl.2017.10.066 |
| [56] |
Hache M, Swoboda KJ, Sethna N, et al. Intrathecal injections in children with spinal muscular atrophy: nusinersen clinical trial experience[J]. J Child Neurol, 2016, 31: 899-906. DOI:10.1177/0883073815627882 |
| [57] |
Hammond SM, Hazell G, Shabanpoor F, et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy[J]. Proc Natl Acad Sci U S A, 2016, 113: 10962-10967. DOI:10.1073/pnas.1605731113 |
| [58] |
Osman EY, Washington Ⅲ CW, Kaifer KA, et al. Optimization of morpholino antisense oligonucleotides targeting the intronic repressor element1 in spinal muscular atrophy[J]. Mol Ther, 2016, 24: 1592-1601. DOI:10.1038/mt.2016.145 |
| [59] |
Zhou H, Janghra N, Mitrpant C, et al. A novel morpholino oligomer targeting ISS-N1 improves rescue of severe spinal muscular atrophy transgenic mice[J]. Hum Gene Ther, 2013, 24: 331-342. DOI:10.1089/hum.2012.211 |
| [60] |
Porensky PN, Mitrpant C, McGovern VL, et al. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse[J]. Hum Mol Genet, 2012, 21: 1625-1638. DOI:10.1093/hmg/ddr600 |