 2022, Vol. 57
2022, Vol. 57



2. 温州医科大学药学院, 浙江 温州 325035
2. School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou 325035, China
肿瘤细胞通过自身或肿瘤微环境过度激活免疫检查点(例如PD-1、CTLA-4、TIM-3等) 通路, 从而导致肿瘤免疫逃逸的发生。其中, PD-1/PD-L1的过度激活对于肿瘤的发展起着至关重要的作用, 阻断PD-1与PD-L1的结合可以逆转肿瘤免疫抑制机制, 有助于提高机体免疫系统杀灭肿瘤的能力[1-5]。目前已有11款PD-1或PD-L1单抗相继上市, 在多种肿瘤的临床治疗中取得了突破性进展, 但抗体类药物普遍存在药源性免疫相关的风险、生产难度大、治疗成本高昂等局限性。相较之下, 针对PD-1/PD-L1通路的小分子药物有望克服抗体药物的上述缺点, 成为单独或联合治疗肿瘤的替代疗法[6]。目前进展最快的是由美国Curis制药公司和印度Aurigene制药公司共同开发的二唑类化合物CA-170, 是一款口服VISTA/PD-L1小分子双重拮抗剂, 也是目前唯一进入临床研究的PD-1/PD-L1小分子抑制剂, 其400 mg剂量组的临床获益率与PD-1/PD-L1抗体相当[7-9]。因此, 寻找新型下调PD-L1的小分子肿瘤免疫抑制剂具有重大意义[10, 11]。
本课题组前期针对免疫检查点PD-L1构建了小分子抑制剂筛选模型, 并对自主构建的具有新颖化学骨架的苦豆碱(1, 图 1) 类生物碱化合物库进行了活性筛选, 发现12N-甲基咪唑磺酰苦豆碱(2, 图 1) 和12N-对三氟甲氧基苯磺酰苦豆碱(3, 图 1) 分别通过溶酶体和蛋白酶体途径下调PD-L1, 在体内外均显示良好的肿瘤免疫治疗效果[12, 13]。但是, 此类化合物下调PD-L1活性的系统构效关系尚未阐明, 其化学结构不同引起下调机制差异的原因有待进一步研究。

|
Figure 1 Structures of aloperine (1), 2, 3 and the modification strategies |

|
Scheme 1 Synthetic route of all the target compounds. Reagents and conditions: (a) R1NCO, CH2Cl2, TEA, rt; (b) Ethyl bromoacetate, EtOH, NaHCO3, reflux; (c) LiOH, H2O, reflux; (d) HOBt, DIEA, EDCI, CH2Cl2, 0 ℃ to rt; (e) Glycine, HOBt, DIEA, EDCI, CH2Cl2, 0 ℃ to rt; (f) HOBt, DIEA, EDCI, CH2Cl2, 0 ℃ to rt; (g) Glycine, H2O, NaOH, 0 ℃ to rt; (h) HOBt, DIEA, EDCI, CH2Cl2, rt |
据此, 本研究集中探讨了在苦豆碱的12N原子上引入全新基团, 如氨甲酰基、氨乙酰基、磺酰氨基乙酰基等对活性的影响, 以此设计合成28个全新的12N取代苦豆碱类衍生物并在高表达PD-L1的乳腺癌MDA-MB-231细胞中评价了其下调PD-L1蛋白水平的活性, 并进一步评价了重点化合物的体外抗肿瘤免疫活性以及作用机制。
结果与讨论 1 化合物的合成目标化合物的合成如合成路线1所示。目标化合物12N-氨甲酰基苦豆碱衍生物4a~4j由1与取代异氰酸酯经亲核加成反应而获得, 总收率为49%~75%。以市售取代胺5a~5g为初始原料, 在碱性条件下, 与溴乙酸乙酯进行烷基化, 然后经酯水解可制备关键中间体N-取代甘氨酸6a~6g, 后者与1在1-羟基苯并三唑(HOBt) 和1-乙基-(3-二甲基氨基丙基) 碳酰二亚胺盐酸盐(EDCI) 作用下发生缩合反应制备12N-氨基乙酰基苦豆碱衍生物7a~7g, 总收率为35%~48%。甘氨酸与1在HOBt和EDCI作用下缩合生成12N-氨基乙酰基苦豆碱I-7h, 再与金刚烷甲酸缩合获得目标物7h, 总收率为45%。取代磺酰氯8a~8j与甘氨酸在碱性下发生缩合反应可制备中间体N-取代磺胺基乙酸9a~9j, 后者与1在HOBt和EDCI作用下缩合生成12N-磺胺基乙酰基苦豆碱衍生物10a~10j, 总收率为39%~53%。所有目标化合物结构经1H NMR、13C NMR以及HR-MS分析确证。目标化合物的收率、理化参数和波谱数据见表 1。
| Table 1 The structures, physical properties and spectral data of the target compounds |
以化合物1和eFT508 (20 μmol·L-1) 为阳性对照[12-14], 采用ELISA方法考察各目标化合物在20 μmol·L-1浓度下与MDA-MB-231细胞共同培养24 h下调PD-L1蛋白水平的作用。目标化合物的结构与活性结果见表 2。
| Table 2 The structures and inhibition rates of PD-L1 level for target compounds. NA: Not active |
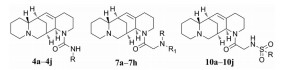



首先在1的12N原子上引入氨甲酰连接基得到10个12N-氨甲酰苦豆碱类似物4a~4j。活性结果显示, 在该连接基末端引入烷基、单取代或双烷基取代苯环, 所得化合物4a~4f下调PD-L1的活性丧失; 而在苯环上引入双氯或三甲基取代, 所得化合物4g、4h和4j下调PD-L1活性大幅度提高, 抑制率分别为35%、47%和34%, 明显优于阳性对照药1和eFT508。将其中一个氯原子替换为三氟甲基, 所得化合物4i的活性显著降低。
将氨甲酰连接基替换为氨乙酰基, 同时保留连接基末端的苯环取代获得8个类似物7a~7h。类似地, 在苯环上引入双氯取代的类似物7g对PD-L1的抑制率为49%, 较单取代苯环类似物7a~7e的活性明显提高。在连接基末端引入大体积的金刚环也有利于活性, 相应化合物7h的抑制率达45%。另外, 在末端伯氨基上引入苯环和甲基双取代, 所得叔胺类似物7f的活性也有大幅度提高, 抑制率为38%。同时, 本研究还在该连接基的末端引入磺酰基, 合成了11个12N-磺酰氨基乙酰基苦豆碱衍生物10a~10j。结果显示, 除了溴噻吩磺酰类似物10j外, 其他类似物的活性均有不同程度的降低。
接下来, 本研究选择抑制率相对较高的7个化合物4g、4h、4j、7f、7g、7h和10j为重点化合物, 利用Western blot方法在同一细胞系上验证了其下调PD-L1的活性(图 2)。蛋白灰度分析数据显示, 阳性药eFT508的抑制率为33%, 4g、7f、7g、7h和10j的抑制率相对较高, 分别为30%、35%、47%、45%和41%, 与上述ELISA结果基本一致。

|
Figure 2 Aloperine derivatives lower PD-L1 protein level. A: MDA-MB-231 cells were treated with the indicated compounds (20 μmol·L-1) for 24 h, the protein level of PD-L1 were measured by Western blot. GAPDH served as the loading control; B: Gray scan results of A. The data presented is the mean ± standard deviation. **P < 0.01, ***P < 0.001 compared with DMSO treatment |
本研究还对上述7个重点化合物开展了细胞毒性评价。选用MDA-MB-231细胞、Vero细胞和293T细胞, 采用MTT测定了各化合物与细胞共培养24 h的半数细胞毒性浓度(CC50)。结果如表 3所示, 化合物7f、7h和10j显示出较高的安全性, 其在Vero和MDA-MB-231细胞中的CC50值均大于200 μmol·L-1。
| Table 3 Cell viability assays of active compounds |
化合物7f、7h和10j均显示出活性高且细胞毒性低的特点, 但是考虑到化合物7h的酯基结构可能导致的代谢不稳定, 选择7f和10j开展进一步研究。
4 化合物7f和10j下调PD-L1总蛋白水平本研究采用Western blot方法考察了7f和10j以不同浓度处理MDA-MB-231细胞时, 下调细胞总PD-L1蛋白水平的量效关系。如图 3A和图 3B所示, 7f和10j均以浓度依赖性的方式下调PD-L1蛋白的表达, 且化合物7f的抑制效率优于10j。同时, 7f下调PD-L1蛋白的作用还具有时间依赖性, 其作用12 h即表现出与作用24 h强度相当的降低PD-L1活性(图 3C)。

|
Figure 3 Compounds down-regulated the expression of total PD-L1 in MB-MDA-231 cells. A: MDA-MB-231 cells were treated with different concentrations of 7f for 24 h; B: MDA-MB-231 cells were treated with different concentrations of 10j for 24 h; C: MDA-MB-231 cells were treated with 7f (20 μmol·L-1) for the different durations. The data presented were the mean ± standard deviation. *P < 0.1, **P < 0.01, ***P < 0.001 and ****P < 0.000 1 compared with DMSO treatment |
PD-1/PD-L1信号通路的激活可抑制T细胞对肿瘤的杀伤作用, 与肿瘤细胞免疫逃逸密切相关, PD-L1水平的降低可激活T细胞对肿瘤细胞的杀伤活性[15]。因此, 本研究评估了7f是否可增强共培养的T细胞对肿瘤细胞的杀伤作用。如图 4A所示, 在MDA-MB-231细胞和Jurkat T细胞(1∶2) 共培养的条件下, 7f可以剂量依赖性方式增强T细胞的杀伤作用。在20 μmol·L-1的浓度下可表现出较好的激活活性(图 4B), 与下调PD-L1表达的起效浓度一致。

|
Figure 4 Compound 7f enhanced the cytotoxicity of T cells. A: MDA-MB-231 cells co-cultured with activated T cells for 24 h with or without different concentrations of 7f pretreatment were subjected to crystal violet staining. The ratio of MDA-MB-231 to T cells is 1∶2; B: The absorbances at 562 nm of the violet dyed crystals in methanol solution were measured. The data presented is the mean ± standard deviation. ***P < 0.001 and ****P < 0.000 1 compared with DMSO treatment |
前期研究显示, 苦豆碱衍生物2和3下调PD-L1水平的机制均不依赖于转录水平, 而是分别通过自噬溶酶体和系统泛素蛋白酶体途径加速其降解[12, 13]。本研究将7f分别与溶酶体抑制剂氯喹(CQ) 和蛋白酶体抑制剂MG132共处理, 以考察7f下调PD-L1水平的作用机制。结果显示(图 5A), MG132与7f共同处理细胞时不影响7f对PD-L1的下调作用, 而CQ与7f的共同作用则可抑制7f对PD-L1蛋白的下调作用, 提示化合物7f主要通过溶酶体途径增强PD-L1的降解, 与2的作用机制一致。

|
Figure 5 Lysosome pathway contributes to 7f-mediated PD-L1 degradation. A: Western blot measuring the PD-L1 expression in MDA-MB-231 cells pre-treated with CQ (50 μmol·L-1) or MG132 (1 μmol·L-1), followed by 7f treatment for 24 h; B: Molecular simulation model of compounds 2, 3 and 7f by Discovery Studio 4.5 |
本研究采用Discovery Studio 4.5软件对化合物2、3和7f开展了分子叠合模拟实验。分子模拟结果(图 5B) 显示, 溶酶体机制的化合物2和7f的正电中心(SO2/CO) 的β位均有一个N原子, 而蛋白酶体机制的化合物3则无此结构特征。溶酶体为酸性环境[16], 化合物2和7f结构中N原子的富电子特征可能有利于其在溶酶体内的聚集, 这可能是其通过溶酶体途径下调PD-L1水平的原因。以上结果可能为未来构建相应作用机制的活性探针解析其直接作用机制奠定基础。
小结本文设计合成了28个全新的12N取代苦豆碱类衍生物, 评价了其在MDA-MB-231细胞中下调PD-L1蛋白水平的活性。其中, 代表性化合物7f表现出良好的活性和较低的细胞毒性。7f以浓度依赖性和时间依赖性的方式下调PD-L1水平, 主要通过调节溶酶体途径促进PD-L1的降解, 从而激活T细胞对肿瘤细胞的杀伤作用, 发挥肿瘤免疫抑制活性。研究结果为苦豆碱衍生物发展成为一类新型的小分子免疫检查点抑制剂提供了科学数据。
实验部分熔点用MP90熔点仪测定(Mettler toledo, Columbus, USA), 未经校正; 1H NMR和13C NMR用Bruker Avance Ⅲ 400、500和600核磁共振仪测定(Varian, San Francisco, USA), 溶剂为DMSO-d6和CDCl3; ESI HR-MS用Autospec Ultima-TOF质谱测定仪测定(Micromass UK Ltd., Manchester, UK); Flash柱分离纯化用Combiflash Rf 200快速制备液相(Teledyne, Nebraska, USA); 紫外检测用ZF-7A手提紫外检测灯(上海宝山顾村电光仪器厂); 薄层色谱(TLC) 采用Merck 60 F254薄层色谱硅胶板(Merck & Co Inc, Darmstadt, German)。吸光度采用酶标仪(Multiskan FC, Thermo, USA) 测定。
所用试剂均为购自百灵威、伊诺凯和通广等试剂公司的分析纯试剂, 未经纯化直接使用。DMEM培养基、RPMI 1640培养基、胎牛血清和青链霉素购自Hyclone (UT, USA)。植物血凝素(PHA)、MG132、CQ和佛波醇12-肉豆蔻酸酯13-乙酸酯(PMA) 购自Sigma (MO, USA)。抗体PD-L1购自Cell Signaling (MA, USA)。GAPDH抗体购自Santa Cruz (CA, USA)。
1 化学合成 1.1 12N-氨基甲酰基苦豆碱4a~4j的合成向苦豆碱(2 mmol) 的无水CH2Cl2 (30 mL) 溶液中缓慢加入异氰酸酯(2 mmol), 在0 ℃下搅拌30 min后, 室温再搅拌30 min。TLC监测反应完全后, 将反应液减压浓缩, 残余物用硅胶匀化, 以CH2Cl2和MeOH为流动相, 经Flash硅胶柱色谱纯化, 得到目标化合物4a~4j。
1.2 12N-氨基乙酰基苦豆碱7a~7g的合成向含取代苯胺5a~5g (5.0 mmol) 和NaHCO3 (6.0 mmol) 的无水乙醇(50 mL) 溶液中加入溴乙酸乙酯(5.5 mmol), 回流8 h。将反应液冷却至室温, 倒入水(30 mL), 并用乙酸乙酯(50 mL) 萃取。分离有机层, 减压浓缩除去溶剂, 将所得的残余物直接加入至含LiOH (5.5 mmol) 的水(40 mL) 溶液中, 回流1.5 h。将反应液冷却至室温后, 用3 mol·L-1盐酸将其pH值调节至2左右, 用CH2Cl2 (20 mL×2) 萃取, 合并有机层并减压浓缩得到中间体6a~6g粗品, 直接用于下一步反应。
在0 ℃条件下, 向上述中间体(2.0 mmol) 的无水CH2Cl2溶液(30 mL) 中依次加入HOBt (2.7 mmol)、二异丙基乙胺(5.2 mL) 和EDCI (4.0 mmol)。搅拌30 min后加入苦豆碱(2.1 mmol), 并转至室温继续搅拌12 h。TLC监测反应完全后, 将该反应液用水(30 mL) 和饱和食盐水(30 mL) 洗涤, 无水Na2SO4干燥, 过滤, 减压浓缩后硅胶匀化, 以CH2Cl2和MeOH为流动相, 经Flash快速硅胶柱色谱分离, 得到目标化合物7a~7g。
1.3 12N-氨基乙酰基苦豆碱7h的合成在0 ℃条件下, 向甘氨酸(2.0 mmol) 的无水CH2Cl2溶液(30 mL) 中依次加入HOBt (2.8 mmol)、二异丙基乙胺(5.4 mL) 和EDCI (4.2 mmol)。搅拌30 min后加入1 (2.2 mmol), 并转至室温继续搅拌12 h。TLC监测反应完全后, 将该反应液用水(30 mL) 和饱和食盐水(30 mL) 洗涤, 无水Na2SO4干燥, 过滤, 减压浓缩得中间体I-7h粗品, 直接用于下一步反应。
将金刚烷甲酸(2.0 mmol) 溶于无水CH2Cl2溶液(30 mL) 中, 在0 ℃条件下, 依次加入HOBt (2.8 mmol)、二异丙基乙胺(5.4 mL) 和EDCI (4.2 mmol)。搅拌30 min后加入上步中间体I-7h, 并转至室温继续搅拌12 h。TLC监测反应完全后, 将该反应液用水(30 mL) 和饱和食盐水(30 mL) 洗涤, 无水Na2SO4干燥, 过滤, 减压浓缩后硅胶匀化, 以CH2Cl2和MeOH为流动相, 经Flash快速硅胶柱色谱分离, 得到目标化合物7h。
1.4 12N-磺酰氨基乙酰基苦豆碱10a~10j的合成在0 ℃条件下, 向甘氨酸(3.0 mmol) 和NaOH (3.6 mol) 的水溶液(15 mL) 中缓慢加入取代磺酰氯8a~8j (3.3 mmol), 然后转至室温, 搅拌3 h。用3 mol·L-1盐酸将该反应液调至pH值2左右, 加入饱和食盐水(30 mL), 用二氯甲烷(30 mL×2) 萃取。合并有机层, 无水Na2SO4干燥, 减压浓缩得到中间体9a~9j, 为白色固体, 不经纯化直接进入下一步反应。
在0 ℃条件下, 向上述白色固体的二氯甲烷溶液(30 mL) 中, 加入HOBt (4.0 mmol)、EDCI (6.0 mmol) 和二异丙基乙胺(1.3 mL)。搅拌30 min后加入苦豆碱(3.0 mmol), 并将该反应转至室温继续搅拌12 h。TLC监测反应完全后, 将该反应液依次用水(50 mL×2) 和饱和食盐水(50 mL) 洗涤, 无水Na2SO4干燥, 过滤, 减压浓缩后硅胶匀化, 以CH2Cl2和MeOH为流动相, 经Flash快速硅胶柱色谱分离, 得到目标化合物10a~10j。
2 细胞培养MDA-MB-231、Jurkat T细胞在RPMI 1640培养基中培养。培养基中补充有10%胎牛血清、100 u·mL-1青霉素和100 μg·mL-1链霉素。猴肾细胞Vero以及人胚肾细胞293T均使用DMEM培养基, 加入10%胎牛血清以及1%青链霉素。5% CO2、37 ℃环境培养。
3 ELISA实验MDA-MB-231细胞以每孔5×104的数量种于24孔板中, 与化合物共培养24 h后按照人PD-L1 ELISA试剂盒(Abcam, ab214565) 说明书方法测定化合物对PD-L1水平的下调作用。
4 免疫印迹分析MDA-MB-231细胞以每孔1×105个种于6孔板中, 与化合物共同培养24 h后, 细胞用冰冷的磷酸盐缓冲盐水(PBS) 洗涤, 并在补充有蛋白酶抑制剂混合物(Sigma P8340) 的RIPA裂解缓冲液中裂解30 min。蛋白通过SDS-PAGE分离并电转移到PVDF膜上。PVDF膜先用适当的一抗孵育, 然后用辣根过氧化物酶(HRP) 偶联的二抗检测。印迹由Tanon 5200系统(Tanon, 上海, 中国) 可视化。
5 MTT法测定细胞毒性取对数期生长的MDA-MB-231细胞、Vero细胞以及293T细胞, 以每孔1×104个细胞的数量接种到96孔板中, 与药物共培养24 h。将20 µL MTT (5 mg·mL-1) 溶液加入到每个孔中, 并于5% CO2、37 ℃培养箱中孵育4 h。去除培养基并加入150 μL DMSO以完全溶解甲瓒结晶后, 测定其在570 nm处的吸光度。
6 T细胞介导的肿瘤细胞杀伤实验使用细胞共培养和结晶紫染色方法进行T细胞介导的肿瘤细胞杀伤实验。Jurkat T细胞由1 μg·mL-1 PHA加50 ng·mL-1 PMA刺激48 h后, 于24孔板中在7f存在下与MDA-MB-231细胞共培养24 h, 通过结晶紫染色观察存活的肿瘤细胞, 后用甲醇溶解结晶, 测定其在562 nm处的吸光度。
7 分子叠合使用Discovery Studio 4.5软件对化合物2、3和7f进行分子叠合实验(molecular overlay)。将Align by filed中的steric和electrostatic参数分别设置为0%和100%。通过比较化合物的三维结构、能量力场性质以及形状性质等特征判断3个分子的叠合效果。
致谢: 目标化合物的核磁共振氢谱、碳谱以及高分辨质谱由中国医学科学院医药生物技术研究所分析测试中心测定。
作者贡献: 张昕彤负责生物部分实验以及初稿的撰写; 王坤和郭志浩负责目标物的合成; 曾庆轩负责文献的调研以及实验方法的设计; 宋丹青对实验过程中出现的问题及论文撰写提供了有益的指导; 本文的通讯作者李迎红副研究员负责实验设计和把关、稿件修改等工作。
利益冲突: 所有作者均声明不存在利益冲突。
| [1] |
Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity[J]. Nature, 2017, 545: 495-499. DOI:10.1038/nature22396 |
| [2] |
Li CW, Lim SO, Xia W, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity[J]. Nat Commun, 2016, 7: 12632. DOI:10.1038/ncomms12632 |
| [3] |
Cha JH, Chan LC, Li CW, et al. Mechanisms controlling PD-L1 expression in cancer[J]. Mol Cell, 2019, 76: 359-370. DOI:10.1016/j.molcel.2019.09.030 |
| [4] |
Chen SM, Crabill GA, Pritchard TS, et al. Mechanisms regulating PD-L1 expression on tumor and immune cells[J]. J Immunother Cancer, 2019, 7: 305. DOI:10.1186/s40425-019-0770-2 |
| [5] |
Yan SJ, Sun L, Wan GH. The mechanism and research progress of drug resistance of PD-1/PD-L1 immunotherapy in tumors[J]. Acta Pharm Sin (药学学报), 2019, 54: 1728-1734. |
| [6] |
Naidoo J, Page DB, Li BT, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies[J]. Ann Oncol, 2015, 26: 2375-2391. DOI:10.1093/annonc/mdv383 |
| [7] |
Sasikumar PG, Sudarshan NS, Adurthi S, et al. PD-1 derived CA-170 is an oral immune checkpoint inhibitor that exhibits preclinical anti-tumor efficacy[J]. Commun Biol, 2021, 4: 699. DOI:10.1038/s42003-021-02191-1 |
| [8] |
Carretero-González A, Lora D, Ghanem I, et al. Analysis of response rate with ANTI PD1/PD-L1 monoclonal antibodies in advanced solid tumors: a meta-analysis of randomized clinical trials[J]. Oncotarget, 2018, 9: 8706-8715. DOI:10.18632/oncotarget.24283 |
| [9] |
Wang FL, Ye WL, Wang S, et al. Discovery of a new inhibitor targeting PD-L1 for cancer immunotherapy[J]. Neoplasia, 2021, 23: 281-293. DOI:10.1016/j.neo.2021.01.001 |
| [10] |
Wang TY, Wu XX, Guo CY, et al. Development of inhibitors of the programmed cell death-1/programmed cell death-ligand 1 signaling pathway[J]. J Med Chem, 2019, 62: 1715-1730. DOI:10.1021/acs.jmedchem.8b00990 |
| [11] |
Pan CH, Yang HY, Lu Y, et al. Recent advance of peptide-based molecules and nonpeptidic small-molecules modulating PD-1/PD-L1 protein-protein interaction or targeting PD-L1 protein degradation[J]. Eur J Med Chem, 2021, 213: 113170. DOI:10.1016/j.ejmech.2021.113170 |
| [12] |
Zhang N, Dou YY, Liu L, et al. SA-49, a novel aloperine derivative, induces MITF-dependent lysosomal degradation of PD-L1[J]. EBioMedicine, 2019, 40: 151-162. DOI:10.1016/j.ebiom.2019.01.054 |
| [13] |
Zeng QX, Wang K, Zhang X, et al. Structure-activity relationship and biological evaluation of 12N-substituted aloperine derivatives as PD-L1 down-regulatory agents through proteasome pathway[J]. Bioorg Chem, 2021, 117: 105432. DOI:10.1016/j.bioorg.2021.105432 |
| [14] |
Chen HM, Li MJ, Ng N, et al. Ruxolitinib reverses checkpoint inhibition by reducing programmed cell death ligand-1 (PD-L1) expression and increases anti-tumour effects of T cells in multiple myeloma[J]. Br J Haematol, 2021, 192: 568-576. DOI:10.1111/bjh.17282 |
| [15] |
Capitani N, Patrussi L, Baldari CT. Nature vs. nurture: the two opposing behaviors of cytotoxic T lymphocytes in the tumor micro environment[J]. Int J Mol Sci, 2021, 22: 11221. DOI:10.3390/ijms222011221 |
| [16] |
Fennelly C, Amaravadi RK. Lysosomal biology in cancer[J]. Methods Mol Biol, 2017, 1594: 293-308. |