 2022, Vol. 57
2022, Vol. 57



动脉粥样硬化形成的血栓性疾病, 例如缺血性脑卒中和心肌梗死是全球发病率和死亡率较高的疾病之一, 血小板的异常活化和聚集是主要原因[1]。抗血小板治疗在临床治疗和预防动脉粥样硬化中有重要意义。目前, COX-1抑制剂阿司匹林、P2Y12拮抗剂氯吡格雷、GPⅡb/Ⅲa受体拮抗剂替罗非班等广泛用于抗血小板治疗[2, 3]。氯吡格雷与阿司匹林联合用药是临床上抗血小板治疗的标准方法, 可以有效抑制血小板的活化, 但同时也影响了正常的止血[4]。为优化抗血小板药物的治疗效果, 减少其出血不良反应, 新型抗血小板药物的研究需求日益凸显。
凝血酶是一种丝氨酸蛋白酶, 目前最强的血小板活化激动剂[5]。它通过与蛋白酶激活受体(protease activated receptor, PARs) 结合发挥作用。目前共发现了PAR1、PAR2、PAR3和PAR4四种PARs受体。其中, 人和哺乳动物血小板表达PAR1和PAR4, 小鼠和大鼠等啮齿动物表达PAR3和PAR4[6]。人体内凝血酶切割PAR1和PAR4的N端, 产生一个新的锁系配体, 从而激活血小板, 使其发挥作用。1999年, 文献[7]报道了人和小鼠的PAR4氨基酸序列同源性约60%。2001年, Sambrano等[8]发现敲除小鼠PAR-4基因会使血小板对凝血酶反应完全丧失。这启发了研究者们可以通过阻断PARs来预防或治疗血栓疾病。除激活PARs受体外, 凝血酶还促进纤维蛋白的形成和蛋白C的激活, 所以PARs受体拮抗剂可以在保留凝血酶其他功能的同时, 阻断凝血酶活化血小板[9, 10]。因此, PARs小分子拮抗剂在抗血小板治疗中可能有更大的治疗指数[11]。
PAR1包含一个类似水蛭素的结构区域, 使其可以以很低的浓度与凝血酶结合, 是凝血酶的高亲和力受体。Vorapaxar (结构见图 1) 是第一个上市的PAR1小分子拮抗剂, 2014年被FDA批准用于预防心肌梗塞和治疗周围动脉疾病[12]。然而, 因为其可增加有中风史或短暂脑缺血发作史的患者颅内出血的风险, 2016年8月默沙东宣布停止在美国继续推广Vorapaxar[13]。随后, PAR4因和PAR1的结构和功能有很大差异而逐渐得到更多的关注。PAR4缺乏水蛭素样凝血酶结合位点, 使得PAR4对凝血酶的敏感度相对较低, 而PAR4在凝血酶裂解位点的下游有一个PAR1不具备的阴离子序列, 该阴离子序列使得PAR4释放更持久的凝血酶信号[14, 15]。PAR4和PAR1在介导凝血酶活化血小板过程中发挥了不同的作用。PAR1激活和驱动快速的初始信号, 而PAR4激活引起较慢但持久的响应[16, 17]。PAR4主要负责维持血小板分泌和血小板促凝功能[18]。PAR4拮抗剂通过抑制血小板活化后期的磷脂酰丝氨酸的暴露和血小板促凝功能预防血栓形成[19]。这些结果表明, PAR4拮抗剂可以抑制血栓形成, 同时出血风险相对PAR1拮抗剂更低。PAR4拮抗剂可能成为一种安全有效的抗血小板药物。

|
Figure 1 Structures of compounds BMS-986120, BMS-986141, vorapaxar |
迄今为止, 已报道的PAR4小分子拮抗剂包括吲唑、吲哚、咪唑并噻二唑、喹啉和喹喔啉衍生物[20]。但只有Bristol-Myers Squibb (BMS) 公司的BMS-986120和BMS-986141 (结构见图 1) 进入了临床试验阶段, 其他化合物均处于临床前研究。2016~2019年, BMS的多篇专利报道了以喹喔啉为结构骨架的衍生物具有抗血小板活性。其中, 先导化合物A是专利WO20171019828中活性最好的结构, 如图 2所示。课题组前期实验中, 保留先导化合物A的喹喔啉环, 然后用氧杂环代替2-甲基得化合物13i, 发现此类结构仍具有抗血小板活性。为继续探究新型PAR4小分子拮抗剂, 本文将8-(二氟甲氧基)-5-芳香基-2, 3-二氢萘[2, 3-g][1,4]二噁英作为母核, 依次探究5位芳香基、8位烷基、氧杂环对活性的影响。基于以上思路设计合成了25个化合物, 并对化合物的生物活性进行评价和构效关系研究。

|
Figure 2 Design of target compounds |
目标化合物的制备方法如路线1所示。以1, 4-苯并二噁烷或1, 3-亚甲二氧基苯作为起始原料经硝化反应得双硝化产物2S、2F, 硝基选择性还原产物再经溴代, 氨基的Boc保护得中间体5S、5F, 然后在三氟乙酸作用下脱去一分子Boc得中间体6S、6F, 中间体6S、6F在碱性条件下与溴乙酸甲酯进行烷基化, 然后在酸性条件下氨基脱保护得中间体8S、8F, 再经二氯化锡还原、关环得中间体9S、9F, 碱性条件下氧化得喹喔啉环, 羟基亲核取代得中间体11S、11F, 中间体11S、11F再经过烷基化得中间体12S、12F, 最后, 中间体11S、11F, 12S、12F与相应的硼酸经Suzuki-Miyaura偶联得目标产物, 反应路线共12步。阳性药BMS-986120参照专利WO2013163279合成, 先导化合物A参照专利WO20171019828合成。

|
Scheme 1 Synthetic route of compounds. Reagents and conditions: (a) 1) c.HNO3, AcOH, rt, 0.5 h; 2) Fuming HNO3, AcOH, 80 ℃, 0.5 h; (b) Fe, AcOH, reflux, 1.5 h; (c) Br2, AcOH, rt, 0.5 h; (d) 4-DMAP, (Boc)2O, THF, rt, 6.0 h; (e) TFA, DCM, rt, 1.0 h; (f) BrCH2COOCH3, Cs2CO3, DMF, rt, 5.0 h; (g) HCl/EA, rt, 3.0 h; (h) SnCl2, c.HCl, MeOH, 70 ℃, 2.5 h; (i) H2O2, NaOH, MeOH/H2O, 85 ℃, 4.5 h; (j) ClF2CCOONa, K2CO3, DMF, reflux, 1.0 h; (k) Ar-B(OH)2, Pd(dppf)Cl2, Na2CO3, toluene/EtOH, reflux, 2.0 h; (l) NaH, R1-OH, THF, rt, 0.5 h |
25个目标化合物经检索均未见文献报道, 并采用1H NMR、13C NMR、MS进行结构确证, 见表 1~3。
| Table 1 Spectral data of compounds |
| Table 2 SAR of 8-(difluoromethoxy)-5-aryl-2, 3-dihydronaphtho[2, 3-b][1,4]dioxine analogues |

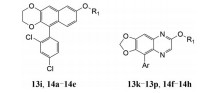
| Table 3 Structures and activities of 13i, 13k-13p, 14a-14h |
以BMS-986120为阳性药, 本实验合成了BMS-986120, 并测得其m. PAR4 AP PRP IC50 (以下简称IC50) 为0.088 μmol·L-1。小鼠动脉血纯化后得到的富血小板血浆(platelet-rich plasma, PRP) 用光电比浊法检测目标化合物对PAR4激动肽(agonist peptide, AP) AYPGKF-NH2诱导的血小板聚集的影响, 测得目标化合物的IC50值如表 2、3。
PAR1和PAR4与G蛋白Gq偶联, Gq在受体激活时会使细胞钙离子外流增加。人类胚胎肾(human embryonic kidney, HEK) 293细胞稳定表达人源PARs, 可用于筛选PARs拮抗剂活性。通过HEK293细胞的荧光成像平板阅读器(fluorescent imaging platereader, FLIPR) 钙流实验可检测化合物对人源PAR1、PAR4的抑制活性, 从而验证代表性化合物对PAR1/PAR4的选择性。测得代表性化合物的PARs FLIPR IC50值如表 4。
| Table 4 In vitro selectivity of BMS-986120 and 13i, 13p, 14a, 14g in the calcium mobilization assays |
用六元氧杂环代替先导化合物A (IC50 = 1.73 μmol·L-1) 的2-甲基, 合成了化合物13i (IC50 = 0.30 μmol·L-1), 化合物13i的活性强于先导化合物A, 说明8-(二氟甲氧基)-5-芳香基-2, 3-二氢萘[2, 3-g][1,4]二噁英类衍生物具有成为新型抗血小板化合物的潜能。为进一步探究该母核的抗血小板活性, 合成了化合物13a~13h、13j, 结构与活性见表 1。首先, Ar选用简单的苯基和吡啶基, 合成了化合物13a (IC50 = 1.89 μmol·L-1)、13b (IC50 > 2.00 μmol·L-1), 活性相对较差, 其中苯环的活性略强于吡啶环。为提高活性, 在苯环上引入不同的取代基。比较化合物13a (IC50 = 1.89 μmol·L-1) 和化合物13c~13g可得, 在苯环上引入合适的基团有利于提高活性, 不同的基团对活性影响较大。特别是化合物13d (IC50 = 0.75 μmol·L-1)和13e (IC50 = 0.88 μmol·L-1), 活性较化合物A分别提高了2.3倍和2.0倍, 说明苯环的2位引入氯原子和甲氧基利于提高活性。化合物13f (IC50 = 1.58 μmol·L-1)、13g (IC50 = 1.36 μmol·L-1) 的活性与化合物A相当, 低于化合物13d和13e, 说明苯环单取代时2位取代较4位取代更利于提高活性。苯环上进行双取代得化合物13h~13j, 结果表明苯环2、4位二氯取代可使活性显著提高, 化合物13i (IC50 = 0.30 μmol·L-1) 较化合物A提高了5.8倍。
固定Ar为2, 4-二氯苯基, 探究R1对活性的影响, 合成了化合物14a~14e。R1的结构及化合物活性见表 2。由化合物13i、14a~14e的活性可得, R1为较小的烷基时更利于提高活性。增大烷基的体积, 如异丙基取代化合物14c、丁基取代化合物14d, IC50都大于2 μmol·L-1, 活性显著降低。另外, 将烷基变为酯基如化合物14e, IC50也大于2 μmol·L-1。最后, 将母核的六元氧杂环变为五元氧杂环, 合成了化合物13k~13q和14f~14h, 结构和活性见表 2。由化合物13a~13c和13k~13m的活性可得, 五元氧杂环的活性与六元氧杂环相当。活性较好的化合物13i (IC50 = 0.30 μmol·L-1)、13p (IC50 = 0.28 μmol·L-1)、14a (IC50 = 0.29 μmol·L-1)、14g (IC50 = 0.26 μmol·L-1) 的IC50见图 3。

|
Figure 3 The IC50 of positive control compound BMS-986120 and several optimized compounds. Data are presented as the mean ± SD (n = 3) |
利用FLIPR钙离子外流实验检测代表性化合物13i、13p、14a和14g对人源PAR1、PAR4的抑制活性, 以验证代表性化合物对PAR1/PAR4的选择性。实验数据如表 4所示。阳性药BMS-986120和代表化合物13i、13p、14a及14g的PAR4 FLIPR IC50值皆在2~7 nmol·L-1, 而PAR1 FLIPR IC50皆大于3 000 nmol·L-1, 表明代表性化合物对人源PAR4有抑制活性, 但对人源PAR1无抑制活性, 说明代表性化合物对PAR4有选择性。
3 小结PAR4拮抗剂可以有效抑制血栓的形成, 同时不易引起出血并发症, 使其可能成为一种安全有效的抗血小板药物。本文设计了一类具有抗血小板活性的新喹喔啉母核, 并合成了25个目标化合物。以处于临床Ⅰ期的BMS-986120为阳性药, 对设计合成的化合物进行抗血小板活性评价、构效关系研究和PAR1/PAR4选择性验证。结果表明, 5位苯基单取代时2位的取代优于4位取代, 当取代基为2-甲氧基苯基和2-氯苯基时活性提高了2.3和2.0倍; 双取代时以2, 4-二氯苯基为最佳, 活性提高了5.8倍。R1为较小的烷基时对活性有利。7-烷氧基-4芳香基-[1, 3]二噁唑[4, 5-g]喹喔啉类衍生物和8-烷氧基-5-芳香基-2, 3-二氢萘[2, 3-b][1,4]二噁英类衍生物的活性相当。最终得活性较好的化合物14a、14g、13i、13p。特别是化合物14g, 活性是先导化合物A的6.7倍, 但其活性只是BMS-986120的十分之三。最后验证了代表性化合物14a、14g、13i、13p的PAR4选择性。本文发现的以2, 3-二氢萘[2, 3-g]二噁唑[1,4]喹喔啉和[1, 3]二噁唑[4, 5-g]喹喔啉为母核的衍生物值得进一步研究, 以开发出高效的新型PAR4选择性拮抗剂。
实验部分实验中所用的试剂及溶剂均为市售分析纯, 1H和13C NMR核磁共振以DMSO-d6或CDCl3为溶剂, 四甲基硅烷(TMS) 为内标, 分别采用Bruker AV-300和Bruker AV-500型核磁共振仪测定。MS采用Agilent 1260-6230 TOF LC-MS液质联用仪测定。薄层色谱(TLC) 采用烟台华阳新材料科技有限公司的HSGF254硅胶板。血小板聚集测定使用北京泰利康信医疗科技有限公司的AG400型半自动血小板聚集仪; ICR小鼠由南京青龙山动物繁殖场或扬州大学实验动物提供中心提供, 动物实验经过中国药科大学实验动物伦理委员会批准; AYPGKF-NH2、SFFLRR-NH2采购于肽佳生物科技有限公司。Tyrode缓冲液采购于雷根生物。除有特别注明外, 所有试剂和溶剂均直接使用。
1 目标化合物的合成6, 7-二硝基-2, 3-二氢苯并[b][1,4]二噁英 (2S) 60%浓硝酸(90 mL) 和冰醋酸(45 mL) 充分搅拌混合后冰浴降至0 ℃, 缓慢滴加化合物2, 3-二氢苯并[b][1,4]二噁英(1S) (20 g, 146.90 mmol), 加入完毕后升至室温反应30 min。TLC监测反应。原料反应完后, 反应液倾入冰水中, 搅拌30 min后抽滤, 滤饼冰水洗涤, 烘干。烘干后的中间体溶于冰醋酸(100 mL), 缓慢小心滴加冰浴冷却后的发烟硝酸(30 mL) 和浓硫酸(4 mL) 的混合溶液。滴加完毕后, 升至室温反应30 min, 后缓慢加热至80 ℃, 剧烈搅拌反应2 h。TLC监测反应。中间体反应完后, 反应液降至室温, 倾入冰水中, 搅拌30 min后抽滤, 滤饼冰水洗涤, 烘干。干燥后的滤饼加入DCM/乙醚的混合溶液加热重结晶, 冷却析晶后过滤得化合物6, 7-二硝基-2, 3-二氢苯并[b][1,4]二恶英(2S) 33 g, 为淡黄色固体, 产率99.3%。中间体(单硝化产物): 1H NMR (300 MHz, CDCl3) δ 7.84~7.76 (m, 2H), 6.95 (d, J = 9.6 Hz, 1H), 4.35 (d, J = 6.0 Hz, 4H)。产物(2S): 1H NMR (300 MHz, CDCl3) δ 7.46 (s, 2H), 4.43 (s, 4H)。类似方法合成化合物2F。1H NMR (300 MHz, CDCl3) δ 7.31 (d, J = 13.8 Hz, 2H), 6.29 (s, 2H)。
7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6-胺 (3S) 氮气气氛下, 化合物6, 7-二硝基-2, 3-二氢苯并[b][1,4]二噁英(2S) (33 g, 145.93 mmol) 溶于冰醋酸(200 mL), 回流30 min, 移除油浴, 分批加入还原铁粉(24.5 g, 3.0 eq)。TLC监测反应。原料反应完后, 反应液冷却至室温后倾入冰水中, 搅拌30 min后过滤产物, 滤饼冰水洗涤。洗涤后的滤饼溶于冰醋酸(200 mL), 回流10 min, 趁热过滤, 滤液倾入冰水中, 搅拌30 min后过滤产物, 滤饼水洗, 干燥, 得化合物7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6-胺(3S) 15.5 g, 为黄色固体, 产率54.1%。1H NMR (300 MHz, CDCl3) δ 7.70 (s, 1H), 6.25 (s, 1H), 4.33 (d, J = 3.8 Hz, 2H), 4.27~4.20 (m, 2H)。类似方法合成化合物3F。1H NMR (300 MHz, CDCl3) δ 7.55 (d, J = 5.6 Hz, 1H), 6.33 (s, 2H), 6.24 (s, 1H), 6.00 (d, J = 5.7 Hz, 2H)。
5-溴7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6-胺 (4S) 冰醋酸(50 mL)中加入液溴(13.9 g, 1.1 eq), 室温下搅拌10 min。化合物7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6-胺(3S) (15.5 g, 79.0 mmol) 溶于冰醋酸(100 mL) 中, 室温下滴加液溴的冰醋酸溶液。滴加完成后室温下再搅拌30 min。TLC监测反应。原料反应完后, 反应液倾入冰水中, 搅拌30 min。抽滤, 滤饼水洗, 烘干, 少量异丙醚打浆, 得化合物5-溴7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6-胺(4S) 18.6 g, 为淡黄色固体, 产率77.2%。1H NMR (300 MHz, CDCl3) δ 7.80 (s, 1H), 6.54 (s, 2H), 4.49 (m, 2H), 4.25 (m, 2H)。类似方法合成化合物4F。1H NMR (300 MHz, CDCl3) δ 7.62 (s 1H), 6.85 (s, 2H), 6.10 (d, J = 3.6 Hz, 2H)。
2, 2'-((5-溴7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6) 氮杂二基) 甲酸叔丁酯 (5S) 室温下, 化合物5-溴7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6-胺(4S) (18.6 g, 62.76 mmol) 和4-二甲氨基吡啶(1.6 g, 0.2 eq) 溶于THF, 加入二碳酸二叔丁酯(36.9 g, 145 mmol), 室温搅拌2.5 h。TLC监测反应。反应完成后, 反应液浓缩, PE∶EA = 3∶1打浆, 得化合物2, 2'-((5-溴7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6) 氮杂二基) 甲酸叔丁酯(5S) 27.9 g。1H NMR (300 MHz, CDCl3) δ 7.73 (s, 1H), 4.50 (dd, J = 5.3, 2.5 Hz, 2H), 4.38 (dd, J = 5.0, 2.7 Hz, 2H), 1.43 (s, 18H)。类似方法合成化合物5F。1H NMR (300 MHz, CDCl3) δ 7.54 (s, 1H), 6.25 (s, 2H), 1.44 (s, 18H)。
(5-溴-7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6-基) 氨基甲酸叔丁酯 (6S) 室温下, 化合物2, 2'-((5-溴7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6) 氮杂二基) 甲酸叔丁酯(5S) 27.9 g溶于二氯甲烷(DCM, 50 mL), 缓慢滴加三氟乙酸(9 mL), 滴加完成后室温下再搅拌10 min。TLC监测反应。反应完成后, 反应液中缓慢加入饱和碳酸氢钠水溶液中和, DCM (100 mL×3) 萃取, 有机相合并, 饱和食盐水洗涤, 无水硫酸钠干燥, 浓缩, 得化合物(5-溴-7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6-基) 氨基甲酸叔丁酯(6S) 31.4 g, 为棕色固体, 无需纯化直接下一步1H NMR (300 MHz, CDCl3) δ 7.62 (s, 1H), 4.50~4.44 (m, 2H), 4.36~4.30 (m, 2H), 1.50 (s, 9H)。类似方法合成化合物6F。1H NMR (300 MHz, CDCl3) δ 7.47 (s, 1H), 6.20 (s, 2H), 1.50 (s, 9H)。
N-((5-溴-7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6-基)-N-(叔丁氧基羰基) 甘氨酸甲酯 (7S) 化合物(5-溴-7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6-基) 氨基甲酸叔丁酯(6S) (27.9 g, 74.36 mmol) 溶于DMF (150 mL), 加入碳酸铯(72.7 g, 3.0 eq), 降至0 ℃, 搅拌10 min, 缓慢滴加溴乙酸甲酯(22.8 g, 2.0 eq), 氮气保护。反应液升至室温搅拌1 h。TLC监测反应。原料反应完后, 加水, EA (100 mL×3) 萃取, 有机相合并, 冰水洗涤3次, 饱和食盐水洗涤, 无水硫酸钠干燥, 浓缩, 异丙醚打浆得化合物N-((5-溴-7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6-基)-N-(叔丁氧基羰基) 甘氨酸甲酯(7S) 棕色固体22.3 g。1H NMR (300 MHz, CDCl3) δ 7.60 (s, 1H), 4.48 (dd, J = 7.6, 3.2 Hz, 2H), 4.42~4.31 (m, 2H), 3.73 (s, 2H), 3.69 (s, 3H), 1.38 (s, 9H)。类似方法合成化合物7F。1H NMR (300 MHz, CDCl3) δ 7.36 (s, 1H), 6.24 (d, J = 14.8 Hz, 2H), 4.32 (s, 2H), 3.72 (s, 3H), 1.47 (d, J = 41.3 Hz, 9H)。
(5-溴-7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6-基) 甘氨酸甲酯 (8S) 化合物N-((5-溴-7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6-基)-N-(叔丁氧基羰基) 甘氨酸甲酯(7S) (21.7 g) 中滴加4 mol·L-1氯化氢的EA (50 mL), 室温搅拌1 h。TLC监测反应。反应完成后, 反应液浓缩, 加入EA复溶后浓缩至干, 异丙醚打浆, 得化合物(5-溴-7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6-基) 甘氨酸甲酯(8S) 橙色固体18.6 g。1H NMR (300 MHz, DMSO-d6) δ 7.67 (s, 1H), 4.53~4.46 (m, 2H), 4.39 (s, 2H), 3.58 (d, J = 2.8 Hz, 3H), 3.33 (s, 2H)。类似方法合成化合物8F。1H NMR (300 MHz, DMSO-d6) δ 7.58 (s, 1H), 6.23 (s, 2H), 4.27 (s, 2H), 3.62 (s, 3H)。
10-溴-2, 3, 8, 9-四氢-[1,4]二噁英[2, 3-g]喹喔啉-7(6H)-酮 (9S) 化合物(5-溴-7-硝基-2, 3-二氢苯并[b][1,4]二噁英-6-基) 甘氨酸甲酯(8S) (18.6 g, 53.58 mmol) 溶于甲醇(150 mL), 滴加浓盐酸(6 mL), 再分批加入二水合氯化亚锡(40.7 g, 4.0 eq), 室温下搅拌10 min, 升温至70 ℃搅拌2.5 h。原料反应完后, 反应液降至室温, 滤液缓慢倒入饱和氢氧化钠溶液中, 加入DCM溶液有二氯化锡化合物析出, 过滤, 萃取, 有机相水洗。得化合物10-溴-2, 3, 8, 9-四氢-[1,4]二噁英[2, 3-g] 喹喔啉-7(6H)-酮(9S), 棕色固体10.0 g。1H NMR (300 MHz, DMSO-d6) δ 10.18 (s, 1H), 6.37 (s, 1H), 5.31 (s, 1H), 4.19 (d, J = 29.2 Hz, 4H), 3.69 (s, 2H)。类似方法合成化合物9F。1H NMR (300 MHz, DMSO-d6) δ 10.19 (s, 1H), 6.44 (s, 1H), 5.95 (s, 2H), 5.38 (s, 1H), 3.67 (s, 2H)。
10-溴-2, 3-二氢-[1,4]二噁英[2, 3-g]喹喔啉-7-醇 (10S) 室温下, 化合物10-溴-2, 3, 8, 9-四氢-[1,4]二噁英[2, 3-g]喹喔啉-7(6H)-酮(9S) 10 g溶于甲醇(50 mL), 加入4.0 mol·L-1的氢氧化钠水溶液(30 mL), 缓慢滴加30%的过氧化氢水溶液(80 mL), 室温下搅拌10 min, 后升至60 ℃搅拌反应20 min。升至85 ℃搅拌反应1 h, 降至室温, 反应液浓缩至小体积, 加水(20 mL), 滴加1 mol·L-1盐酸调节pH = 2~3, 冰浴降温至0 ℃搅拌20 min, 抽滤, 滤饼水洗, 少量丙酮∶乙醚= 1∶1的混合溶液洗涤, 烘干, 得化合物10-溴-2, 3-二氢-[1,4]二噁英[2, 3-g]喹喔啉-7-醇(10S) 白色固体10 g。1H NMR (300 MHz, DMSO-d6) δ 12.31 (s, 1H), 8.07 (s, 1H), 6.77 (s, 1H), 4.39 (d, 4H)。类似方法合成化合物10F。1H NMR (300 MHz, DMSO-d6) δ 8.07 (s, 1H), 6.79 (s, 1H), 6.26 (s, 2H)。
5-溴-8-(二氟甲氧基)-2, 3-二氢-[1,4]二氧杂[2, 3-g]喹喔啉 (11S) 化合物10-溴-2, 3-二氢-[1,4]二噁英[2, 3-g]喹喔啉-7-醇(10S) (10.0 g, 35.33 mmol) 和无水碳酸钾(29.3 g) 溶于DMF (100 mL), 100 ℃下搅拌反应5 min, 分批加入二氟氯乙酸钠(26.9 g), 100 ℃下搅拌反应20 min。TLC监测反应。原料反应完后, 反应液降至室温, 加水稀释, EA (20 mL×3) 萃取, 有机相合并, 冰水洗涤3次, 饱和食盐水洗, 无水硫酸钠干燥, 浓缩, 硅胶柱色谱(PE∶DCM = 10∶1-2∶1), 异丙醚打浆, 得化合物5-溴-8-(二氟甲氧基)-2, 3-二氢-[1,4]二氧杂[2, 3-g]喹喔啉(11S) 600 mg, 白色固体, 产率51.2%。1H NMR (300 MHz, DMSO-d6) δ 8.70 (s, 1H), 7.81 (t, JHF = 71.7 Hz, 1H), 7.36 (s, 1H), 4.54 (dd, J = 5.2, 2.2 Hz, 2H), 4.50~4.44 (m, 2H)。类似方法合成化合物11F。1H NMR (300 MHz, DMSO-d6) δ 8.68 (s, 1H), 7.80 (t, JHF = 71.7 Hz, 1H), 7.35 (s, 1H), 6.42 (s, 2H)。
5-溴-8-烷氧基-2, 3-二氢-[1,4]二氧杂[2, 3-g]喹喔啉 (12S) 合成通法 冰浴加入60%氢化钠和相应的醇(2 mL) 溶于DMF, 室温下搅拌30 min, 再加入5-溴-8-(二氟甲氧基)-2, 3-二氢-[1,4]二氧杂[2, 3-g]喹喔啉(11S) 0.5 g, 室温反应, TLC监控反应。原料反应完后, 加水稀释, EA (20 mL×3) 萃取, 有机相合并, 冰水洗涤3次, 饱和食盐水洗, 无水硫酸钠干燥, 硅胶柱色谱分离。产率70%~80%, 皆为白色固体。
目标化合物合成通法 化合物12Sa~12Sf、12Fa、12Ff (0.5 g, 1.0 eq) 和不同的硼酸(4.0 eq) 溶于甲苯(3 mL) 和乙醇(1 mL), 严格氮气保护, 加入[1, 1′-双(二苯基膦基) 二茂铁] 二氯化钯(0.02 eq) 和2 mol·L-1碳酸钠水溶液(3.0 eq), 升温回流反应30 min~12 h, TLC监测反应。反应完后, 反应液降至室温, 加EA/H2O分液, 有机相无水硫酸钠干燥, 浓缩, 硅胶柱色谱得目标化合物, 少量异丙醚打浆后得到纯品。产率40%~70%, 皆为白色固体。
2 生物活性测试 2.1 小鼠PRP制备健康ICR小鼠腹腔注射5%水合氯醛, 以6 mL·kg-1的剂量麻醉大约5~10 min。此时小鼠呼吸变缓, 呼吸方式以腹式呼吸为主, 肌肉不再紧绷且眼睑反射消失, 对严重刺激都不产生有害反射, 表示小鼠已经麻醉。将麻醉好的小鼠固定在纸板上, 沿着腹部中央剪开皮肤与腹膜, 使腹腔暴露。左手轻柔地将小鼠内脏剥到左侧, 暴露腹部主动脉。右手持吸有0.1 mL抗凝剂的2.5 mL注射器, 针孔朝下30°角刺入腹主动脉, 然后将针头放平, 右手固定针管和针头不动, 左手缓缓拉注射器活塞, 使得血液缓慢进入注射器中, 直到有阻力使其停止。拔出注射器, 轻柔上下颠匀注射管中的血液和抗凝剂。将全血收集于抗凝剂润洗过的15 mL硅烷化管中, 并置于37 ℃水浴锅中保温, 直至血样采集完毕。小鼠全血用生理盐水稀释(血/生理盐水= 3∶1) 后, 1 080 r·min-1离心10 min, 取上层血浆即PRP (platelet-rich plasma) 于生理盐水润洗过的硅烷化管, 37 ℃水浴静置备用。吸取PRP后剩余部分3 000 r·min-1离心5 min, 上层即为PPP (platelet-poor plasma), 吸取PPP于生理盐水润洗后的15 mL硅烷化管中, 37 ℃水浴静置备用。
2.2 血小板聚集实验吸取300 μL Tyrode, s buffer于干净的测试杯中, 置于血小板聚集测试区调零(仪器需要提前预热至37 ℃)。再准确吸取270 μL PRP (已用PPP调整血小板数量级每毫升3×108个) 悬液于预热槽中, 分别加入20 μL生理盐水或者不同浓度的待测样品, 37 ℃孵育5 min后置于测试区, 向测试杯中依次加入磁珠和10 μL AYPGKF-NH2诱导剂后立即开始测试其聚集率, 最终聚集仪显示5 min内血小板的最大聚集率。抑制率(%) = [(阴性组血小板最大聚集率-样品组血小板最大聚集)/阴性组血小板最大聚集率]。使用软件Graphpad Prism 7.0, 以不同浓度下样品抑制率计算IC50。
2.3 HEK293细胞的荧光成像平板阅读器(FLIPR)钙流检测实验HEK293/ga15/PAR1和a PAR1 AP (SFFLRR-NH2) 用于检测PAR1的抑制活性。HEK293/ga15/PAR4和a PAR4 AP (AYPGKF-NH2) 用于检测PAR4的抑制活性。将培育好的细胞基质液按每孔30 mL加入至384孔透明底黑板中, 30 min后离心1 min。用细胞解离液消化HEK293/Ga15/PARs细胞。然后每孔45 000个细胞铺板, 37 ℃、5% CO2孵育24 h。孵育完成后, 去除培养基将配置好的缓冲液以每孔100 μL的量迅速加入, 37 ℃、5% CO2避光继续孵育60 min。将样品化合物溶解在DMSO中, 用HBSS缓冲液将浓度稀释到4%, 再每孔添加25 μL稀释液, 空白组加入25 μL的HBSS, 室温下避光孵育15 min。孵育完成后, 将384孔板置于FlexStation读板仪(Molecular Devices Co., USA) 上, 每孔加入25 μL的PAR AP溶液, 测定各孔的相对荧光强度, 计算样品化合物的抑制率。通过测定不同浓度下的抑制率, 可通过GraphPad Prism 7.0进行数据处理, 计算其IC50。
作者贡献: 所有作者都对化合物的设计做出了贡献; 朱雄是本文的通讯作者; 谢柔洁是本文的第一作者, 负责实施实验、整理数据及撰写稿件; 刘尚德负责提出研究思路、设计实验方案和整体把关、修改稿件; 袁铎和李杉杉负责指导稿件的撰写和修改。
利益冲突: 本文的研究内容无任何利益冲突。
| [1] |
Michelson, Lan DA. Antiplatelet therapies for the treatment of cardiovascular disease[J]. Nat Rev Drug Discov, 2010, 9: 154. DOI:10.1038/nrd2957 |
| [2] |
Franchi F, Angiolillo DJ. Novel antiplatelet agents in acute coronary syndrome[J]. Nat Rev Cardiol, 2015, 12: 30-47. DOI:10.1038/nrcardio.2014.156 |
| [3] |
Husted S, Emanuelsson H, Heptinstall S, et al. Pharmacodynamics, pharmacokinetics, and safety of the oral reversible P2Y12 antagonist AZD6140 with aspirin in patients with atherosclerosis: a double-blind comparison to clopidogrel with aspirin[J]. Eur Heart J, 2006, 27: 1038-1047. DOI:10.1093/eurheartj/ehi754 |
| [4] |
Li SS, Zhu X. Research progress on protease-activated receptor antagonists in the field of antiplatelet[J]. J Pharm Res (药学研究), 2019, 38: 543-549. |
| [5] |
Connolly TM, Condra C, Feng DM, et al. Species variability in platelet and other cellular responsiveness to thrombin receptor-derived peptides[J]. J Thromb Haemost, 1994, 72: 627-633. DOI:10.1055/s-0038-1648926 |
| [6] |
Gemma V, Manuel G, Monika A, et al. Intracellular platelet signalling as a target for drug development[J]. Vasc Pharmacol, 2018, 111: 22-25. DOI:10.1016/j.vph.2018.08.007 |
| [7] |
Coughlin SR. How the protease thrombin talks to cells[J]. Proc Natl Acad Sci U S A, 1999, 96: 11023-11027. DOI:10.1073/pnas.96.20.11023 |
| [8] |
Sambrano GR, Weiss EJ, Zheng YW, et al. Role of thrombin signalling in platelets in haemostasis and thrombosis[J]. Nature, 2001, 413: 74-78. DOI:10.1038/35092573 |
| [9] |
Judge HM, Jennings LK, Moliterno DJ, et al. PAR1 antagonists inhibit thrombin-induced platelet activation whilst leaving the PAR4-mediated response intact[J]. Platelets, 2015, 26: 236. DOI:10.3109/09537104.2014.902924 |
| [10] |
French SL, Arthur JF, Lee H, et al. Inhibition of protease‐activated receptor 4 impairs platelet procoagulant activity during thrombus formation in human blood[J]. J Thromb Haemost, 2016, 14: 1642-1654. DOI:10.1111/jth.13293 |
| [11] |
Bulani Y, Sharma S. Therapeutic potential of targeting protease activated receptors in cardiovascular diseases[J]. Curr Pharm Design, 2015, 21: 4392-4399. DOI:10.2174/138161282130151007144725 |
| [12] |
Poole RM, Elkinson S. Vorapaxar: first global approval[J]. Drugs, 2014, 74: 1153-1163. DOI:10.1007/s40265-014-0252-2 |
| [13] |
Vranckx P, White HD, Huang Z, et al. Validation of BARC bleeding criteria in patients with acute coronary syndromes[J]. J Am Coll Cardiol, 2016, 67: 2135-2144. |
| [14] |
Covic L, Gresser AL, Kuliopulos A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets[J]. Biochemistry, 2000, 39: 5458-5467. DOI:10.1021/bi9927078 |
| [15] |
Hosokawa K, Ohnishi T, Miura N, et al. Antithrombotic effects of PAR1 and PAR4 antagonists evaluated under flow and static conditions[J]. Thromb Res, 2014, 133: 66-72. DOI:10.1016/j.thromres.2013.10.037 |
| [16] |
Wong PC, Seiffert D, Bird JE, et al. Blockade of protease-activated receptor-4 (PAR4) provides robust antithrombotic activity with low bleeding[J]. SciTransl Med, 2017, 9: f5294. |
| [17] |
Covic L, Singh C, Smith H, et al. Role of the PAR4 thrombin receptor in stabilizing platelet-platelet aggregates as revealed by a patient with Hermansky-Pudlak syndrome[J]. J Thromb Haemost, 2002, 87: 722-727. DOI:10.1055/s-0037-1613071 |
| [18] |
Jacques SL, Kuliopulos A. Protease-activated receptor-4 uses dual prolines and an anionic retention motif for thrombin recognition and cleavage[J]. Biochem J, 2003, 376: 733. DOI:10.1042/bj20030954 |
| [19] |
Su X, Su W, He Z, et al. Tripeptide SQL inhibits platelet aggregation and thrombus formation by affecting PI3K/Akt signaling[J]. Cardiovasc Pharmacol, 2015, 66: 254-260. DOI:10.1097/FJC.0000000000000269 |
| [20] |
Liu S, Li S, Yuan D, et al. Protease activated receptor 4 (PAR4) antagonists: research progress on small molecules in the field of antiplatelet agents[J]. Eur J Med Chem, 2020, 209: 112893. |