 2021, Vol. 56
2021, Vol. 56



耐药菌尤其革兰阴性(G-) 耐药菌引起的感染成为全球严重的公共健康问题, 但目前尚未有针对G-耐药菌的有效治疗药物用于临床。氨曲南(aztreonam, 1, 图 1), 作为第一个临床上使用的单环β内酰胺类抗生素, 于1986年由美国FDA批准用于抗G-菌的临床治疗, 包括尿路感染、下呼吸道感染、血液感染、皮肤软组织感染、腹膜炎等腹腔感染以及生殖道感染[1]。氨曲南通过和细菌的青霉素结合蛋白(PBPs) 发生共价结合而发挥作用, 对细菌产生的质粒介导和大部分染色体介导的β内酰胺酶(如MBLs) 高度稳定, 具有肾毒性低, 免疫原性弱, 与青霉素类、头孢菌素类交叉过敏少等特点, 因此可用于治疗肾功能损害患者以及耐药革兰阴性菌感染[1, 2]。但是, 氨曲南在胃肠道以离子形式存在, 几乎不吸收, 口服生物利用度极低[2], 临床上一般采用静脉输注方式给药。为拓展氨曲南的使用范围, 本研究拟通过制备氨曲南前药来探索其作为口服制剂的可能性, 具有重要意义。

|
Figure 1 Structures of compounds 1-5 and aztreonam prodrugs |
前药策略可提高口服生物利用度, 降低不良反应[3, 4], 在β内酰胺类抗生素的前药设计中得到广泛应用。如图 1所示, 将静脉给药的部分青霉素、头孢菌素类和碳青霉烯类药物结构中的羧基转变成羧酸酯以制成相应的羧酸酯前药, 可明显改善原药的药动学特征以及口服生物利用度[5]; 进而被体内广泛存在的酯酶水解成羧酸原药以发挥抗菌作用, 如匹氨西林(2)、头孢泊肟酯(3) 以及替比培南伏匹酯(4) 等。Gordon等[6, 7]通过氨曲南的磺酸基引入磺酸酯基制备了一系列前药化合物, 代表性化合物5可通过与羧酸酯酶1 (CES1) 水解而释放氨曲南原药。
基于此, 本研究拟通过氨曲南的羧酸构建羧酸酯类前药, 以氨曲南为母药, 参考其ClopP值, 以羧基为把手引入不同结构类型的支链烷基酯、直链烷基酯以及双酯, 共设计、合成了9个氨曲南羧酸酯前药, 评价了其体外抗菌活性、细胞毒性、体外药代动力学性质, 并选取重点化合物开展了体内药代动力学评价。
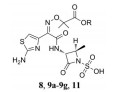
结果与讨论 1 化合物的合成目标化合物氨曲南叔丁酯8的合成见合成路线1, 由头孢他啶活性酯6和氨曲南母核7经缩合反应获得。对比了6种不同的缩合剂对反应产率的影响, 如2-(7-氮杂苯并三氮唑)-N, N, N', N'-四甲基脲六氟磷酸酯(HATU)、二环己基碳二亚胺(DCC)、二异丙基碳二亚胺(DIC)、1-乙基-3(3-二甲基丙胺) 碳二亚胺(EDCI)、2-氯-4, 6-二甲氧基-1, 3, 5-三嗪(CDMT) 和2-氯-4, 6-二甲氧基-1, 3, 5-三嗪(COMU)。其中, 以DIC为缩合剂时反应收率最为理想。经不同碱性试剂及配伍比例的正交实验结果表明, 1.0 eq的6和1.4 eq的7在1.5 eq DIC、3.0 eq三乙胺(TEA) 和催化量4-二甲氨基吡啶(DMAP) 的共同作用下, 可以通过重结晶以27%产率获得目标化合物。
氨曲南与不同直链一级醇发生缩合反应可以制备目标产物9a~9g (合成路线2)。以DIC为缩合剂, 在甲醇或乙醇中利用二次重结晶获得目标产物9a和9b, 收率为25%~28%。但是, 该缩合条件不适合用于制备其同系物9c~9g, 反应收率大为降低。因此, 本研究对比了其他的缩合剂(组合), 如EDCI、EDCI/1-羟基苯并三唑(HOBT)、N, N-羰基-二咪唑(CDI) 或CDMT与碱性试剂N-甲基吗啉(NMM)、N, N-二异丙基乙胺(DIEA) 配伍等对反应的影响, 筛选出最优的反应条件, 即以相应醇为溶剂, EDCI和HOBT与DIEA配伍, 可以14%~28%的收率收获目标化合物9c~9g。氨曲南与化合物10 (特戊酸氯甲酯) 在碳酸钾的作用下, 发生缩合反应得到化合物11 (合成路线2)。

|
Scheme 1 Synthetic route of target compound 8 |

|
Scheme 2 Synthetic route of target compounds 9a-9g and 11 |
合成的目标化合物的理化参数和波谱数据见表 1。
| Table 1 The structures, physical properties and spectra data of target aztreonam prodrugs |
以氨曲南为对照药物, 测定9个目标前药对不同革兰阴性菌的体外抗菌活性, 菌株包括ATCC及中国医院临床分离的大肠杆菌(Escherichia coli)、阴沟肠杆菌(Enterobacter cloacae)、产气肠杆菌(Enterobacter aerogenes)、鲍曼不动杆菌(Acinetobacter baumannii)、铜绿假单胞菌(Pseudomonas aeruginosa) 以及耐药肺炎克雷伯菌(Klebsiella Pneumoniae) 等。如表 2所示, 它们的抗菌活性均消失, 说明了羧基是抗菌活性的必需基团, 裸露的羧基有利于活性。
| Table 2 Antibacterial activities of the target aztreonam prodrugs. *MIC was determined by agar dilution; aEscherichia coli; bKlebsiella Pneumoniae; cEnterobacter cloacae; dEnterobacter aerogenes; ePseudomonas aeruginosa; fAcinetobacter baumannii |
以氨曲南为对照化合物, 在3种细胞(HepG 2细胞、Vero细胞、293T细胞) 模型上评价了所有前药化合物的细胞毒性。测定结果如表 4所示, 所有前药化合物在上述细胞中的CC50值均高于0.32 mmol·L-1, 其安全性优于或与氨曲南本身相当。
| Table 3 Hydrolysis rate of aztreonam prodrugs in different plasmas. aPropantheline bromide; bEnalapril maleate salt; cBisacodyl; dProcaine hydrochlorid |
| Table 4 Cytotoxicity of the target compounds |
分别以含有羧酸酯结构的前药普鲁本辛(propantheline) 和依那普利(enalapril) 为对照化合物, 采用HPLC-MS/MS方法测定了氨曲南前药化合物在啮齿类动物(小鼠和大鼠) 血浆中的稳定性(图 2和表 3)。结果显示, 所有前药化合物均可以被小鼠或大鼠血浆酯酶迅速且完全代谢并释放原药, 其在30 min内均可被全部水解, 并且此类化合物在大鼠血浆中的酶解速率可能要略高于小鼠。

|
Figure 2 The stability of target aztreonam prodrugs in mouse and rat plasmas |
动物血浆中的酯酶的种类和含量存在种属差异, 例如CES仅在啮齿类动物, 如小鼠和大鼠的血浆中表达, 而在非啮齿类动物的血浆中表达量最高的酯酶则是氧磷酶(PON) 和丁酰胆碱酯酶(BChE)[8, 9]。本研究进一步考察了上述前药化合物在非啮齿类动物(犬、猴和人) 血浆中的代谢稳定性, 分别以羧酸酯前药比沙可啶(bisacodyl)、普鲁卡因(procaine) 和普鲁本辛为对照化合物。结果显示(图 3和表 3), 所有前药化合物在非啮齿类动物血浆中的酶解速率均明显减慢, 尤其是氨曲南单酯前药9a~9g在30 min内的水解率均不超过60%, 在120 min内的水解率均不超过80%。提示基于单环β内酰胺的氨曲南单酯可能对CES高度敏感, 而对其他酯酶的敏感性大为降低。相对的, 氨曲南双酯前药11在人和猴血浆中的酶解速率相对较高, 120 min内的水解率达到99.3%和83.5%, 这可能与双酯结构更易被灵长类动物的血浆酯酶识别的机制有关[8]。

|
Figure 3 The stability of target aztreonam prodrugs in dog, monkey and human plasmas |
进一步对氨曲南单酯前药酶解速率的种属差异进行了分析。当化合物的ClogP值小于2时(9a~9c), 其在犬、猴和人血浆中的酶解速率并无显著差异, 120 min时的水解率均在35%以内。随着化合物ClogP的逐渐增加, 其在3个种属血浆中的酶解速率出现了显著差异, 且酶解速率的顺序为人 > 猴 > 犬。其中, ClogP值最大的化合物9g在120 min时在人、猴和犬血浆中的水解率分别为55.8%、31.9%和14.1%。
此外, 还发现除9a以外, 所有的氨曲南单酯前药在人血浆中的水解速率还与载体的长度相关, 随着载体链长的增加, 其代谢速度也随之增加。乙酯9b、正丙酯9c、正丁酯9d、正戊酯9e、正己酯9f和正辛酯9g在120 min时的水解率分别为9.7%、19.6%、31.5%、45.6%、47.6%和55.8%。同时, 考察了烷基链的支链或直链状态对水解速率可能产生的影响, 将化合物9d的正丁酯替换为叔丁酯, 所得的前药8的水解速率与9d并没有显著差异。据此推测氨曲南单酯类前药在人血浆中的酶解速率可能与化合物本身的ClogP值呈正相关。
ClogP值与药物的口服吸收性质关系密切, 为了进一步考察氨曲南前药化合物的代谢行为与其ClogP值之间的关系, 本研究拟选择化学性质较为稳定的单酯化合物[8] 9f和9g为代表性化合物, 开展体内药代动力学评价。
5 代表性前药化合物药代动力学评价如图 4和表 5所示, 以雄性SD大鼠为模型, 以25 mg·kg-1的剂量一次性口服给药9f和9g后, 利用HPLC-MS/MS技术同时检测血浆中前药本身和氨曲南的含量。结果显示, 两组大鼠血浆中均不能检测到前药本身, 进一步证明了氨曲南羧酸酯对大鼠血浆酯酶的高度敏感性。其中, 前药9g组释放氨曲南的最高血药浓度为96.3 ng·mL-1, 浓度-时间曲线下面积(AUC0-inf) 为364 ng·h·mL-1, 明显高于9f, 同时9g组氨曲南的半衰期(t1/2) 较9f也有明显提升, 提示其具有相对较高的口服吸收, 而这一结果与其相对较高的ClogP值相匹配。氨曲南在健康受试者身上, 以500 mg的剂量一次性口服给药氨曲南后检测到其16 h内的AUC0-16为0.45 μg·h·mL-1, 其口服生物利用度为约0.55%[10]。虽然化合物9g的口服药代动力学性质尚不理想, 但研究结果提示其可以增加氨曲南的口服吸收。因此, 通过改变酯侧链的结构提高与改善氨曲南的口服药代特性与生物利用度的策略是可行的。

|
Figure 4 Plasma concentration of aztreonam after dosing of prodrugs 9f and 9g |
| Table 5 Pharmacokinetics of aztreonam prodrugs. The data listed in this table was referred to aztreonam |
与在氨曲南的磺酸基上引入前药载体相比[7], 在氨曲南的羧基上引入酯类载体合成难度明显降低, 可操作性强。此外, Gordon等[7]仅测试了氨曲南磺酸酯前药5被单一羧酸酯酶CES1水解的情况, 其在人整体血浆中的稳定性尚不可知。本研究发现的前药化合物9g在人血浆中2 h内的水解率为55.8%, 从开发长效的口服氨曲南前药的角度考虑, 其水解速率在合理范围内。
小结本研究设计合成并评价了9个氨曲南酯类化合物, 评价了其在不同种属动物血浆中的酯酶稳定性, 并选取重点化合物进行了体内主要药代参数的测定。结果表明, 氨曲南羧酸酯对血浆酯酶的敏感性存在种属差异, 其在小鼠、大鼠血浆中的酶解速率远远高于犬、猴和人, 且在人血浆内的酶解速率与其ClogP值可能呈正相关。上述研究结果提示, 在对酯类前药化合物开展药代动力学评价时, 可能需要充分考虑到种属差异对其药代性质产生的影响。此研究对单环β内酰胺抗生素前药的开发研究具有一定的参考价值。
实验部分 1 化学合成熔点用CXM-300型精密熔点仪测定, 温度未校正; 核磁共振氢谱和碳谱分别采用Varian Mercury Plus 400 (600 MHz) 和Bruker Avance Ⅲ 500 (151 MHz) 核磁共振仪测定, 溶剂均为DMSO-d6, TMS为内标; HR-MS用AutospecUltima-TOF质谱测定仪测定; Flash柱分离纯化用Combiflash Rf200快速制备液相; 荧光检测用ZF-20D暗箱式紫外分析仪; 薄层色谱(TLC) 采用E-Merck公司预铺硅胶铝箔卷。所用试剂为Acros公司和国产分析纯或化学纯的试剂。
1.1 氨曲南叔丁酯(8)室温下, 将(Z)-2-[(2-氨基噻唑-4-基)-(苯并噻唑-2-基硫基羰基) 亚甲胺基氧基]-2-甲基丙酸叔丁酯(6, 0.50 g, 1.5 mmol) 溶于少量的DMF (5 mL) 中, 依次加入TEA (0.32 g, 3.15 mmol)、trans-3(S)-氨基-4-甲基-2-氧代-1-氮杂环丁烷基磺酸(7, 0.38 g, 2.1 mmol)、DIC (0.28 g, 2.25 mmol)、HOBT (0.30 g, 2.25 mmol) 以及DMAP (0.018 g, 0.15 mmol), 继续反应18 h, TLC检测反应完全。加少量硅胶匀化样品, 以石油醚/乙酸乙酯和二氯甲烷/甲醇为流动相在25 g硅胶柱上进行Flash分离纯化。将收集到的组分溶于乙醇(2 mL), 用盐酸/乙醇溶液调pH值至2.5~3.0, -20 ℃下静置48 h, 抽滤得到白色固体8 (0.2 g, 27%)。
1.2 氨曲南甲酯(9a)室温下, 将氨曲南(0.50 g, 1.15 mmol) 溶于甲醇(15 mL) 中, 之后依次加入TEA (0.15 g, 1.48 mmol)、DIC (0.19 g, 1.5 mmol) 以及DMAP (0.004 g, 0.033 mmol), 继续反应48 h, TLC检测反应完全。室温下用盐酸乙醇溶液将反应体系pH调至2.5~3.0, 并于-20 ℃下养晶48 h。抽滤, 将得到的固体在室温下用少量的DMSO (2 mL) 溶解, 之后加入约10倍体积的乙醇(20 mL), 保温-20 ℃养晶48 h, 抽滤得到白色固体9a (0.13 g, 25%)。
1.3 氨曲南乙酯(9b)以氨曲南为起始原料, 乙醇作为反应溶剂, 参照化合物9a的制备方法, 得到白色固体9b (0.15 g, 28%)。
1.4 氨曲南正丙酯(9c)室温下, 将氨曲南(0.65 g, 1.5 mmol) 溶于正丙醇(15 mL) 中, 之后依次加入DIEA (0.37 g, 2.9 mmol)、EDCI (0.33 g, 1.7 mmol) 以及HOBT (0.23 g, 1.7 mmol), 继续反应48 h, TLC检测反应完全。加少量硅胶匀化样品, 以石油醚/乙酸乙酯和二氯甲烷/甲醇为流动相在12 g硅胶柱进行Flash分离纯化。将收集到的组分溶于蒸馏水(2 mL), 并用稀盐酸调pH值至2.0, 抽滤得淡黄色固体, 冷丙酮(10 mL) 精制得白色固体9c (0.10 g, 14%)。
1.5 氨曲南正丁酯(9d)以氨曲南为起始原料, 正丁醇作为反应溶剂, 参照化合物9c的制备方法, 得到白色固体9d (0.16 g, 22%)。
1.6 氨曲南正戊酯(9e)以氨曲南为起始原料, 正戊醇作为反应溶剂, 参照化合物9c的制备方法, 得到白色固体9e (0.21 g, 28%)。
1.7 氨曲南正己酯(9f)以氨曲南为起始原料, 正己醇作为反应溶剂, 参照化合物9c的制备方法, 得到白色固体9f (0.12 g, 15%)。
1.8 氨曲南正辛酯(9g)以氨曲南为起始原料, 正辛醇作为反应溶剂, 参照化合物9c的制备方法, 得到白色固体9g (0.14 g, 17%)。
1.9 氨曲南特戊酸甲酯(11)室温下, 将氨曲南(1.0 g, 2.3 mmol) 溶于DMF (10 mL) 当中, 之后加入特戊酸氯甲酯(10, 0.7 g, 4.6 mmol) 以及碳酸钾(0.6 g, 4.6 mmol), 室温下继续反应6 h, TLC检测反应完全。加少量硅胶匀化样品, 以石油醚/乙酸乙酯和二氯甲烷/甲醇为流动相在25 g硅胶柱上进行Flash分离纯化。将收集的目标组分用蒸馏水(4 mL) 溶解, 并用稀盐酸调pH值至2.0, 抽滤得淡黄色固体, 冷丙酮(15 mL) 精制得白色固体11 (0.1 g, 8%)。
2 抗菌活性的测定本研究按照CLSI/NCCLS标准, 采用平皿琼脂二倍稀释法及Denlay多点接种器(multipoint inoculator A400) 进行抑菌活性测定试验, 用水解酪蛋白胨(Mueller-Hinton agar) 肉汤及脑心浸液扩增试验菌株。本研究使用的菌株来源于ATCC以及中国医院临床分离的细菌菌株, 以氨曲南为阳性对照药。将药物用水解酪蛋白肉汤稀释成128、64、32、16、8、4、2、1、0.5、0.25、0.125、0.06和0.03 μg·mL-1等浓度, 分别置于平皿中。等MH琼脂培养基溶化后, 再定量吸取将其加入含待测药物样品的平皿内, 充分混匀。实验菌株接种后, 于35 ℃恒温培养18 h, 观察菌株生长情况, 无菌生长的平皿中所含药物浓度就是测得的最低抑菌浓度(MIC)。
3 细胞毒性测定采用CCK8法进行。取对数生长期细胞, 消化后计数, 将细胞接种于96孔培养板。培养过夜待贴壁后, 用无血清培养基稀释药物, 用浓度梯度的药物处理细胞。72 h后, 去除培养液, 加入10倍稀释的CCK8试剂100 μL, 37 ℃温箱孵育2~3 h后, 震荡3 min混匀, 用酶标仪测定450 nm吸收度值(A)。细胞存活率%=(加药细胞A-本底A) / (对照细胞A-本底A)×100%。每检测点取3个平行孔的平均值, 绘制抑制曲线。
4 血浆酯酶代谢稳定性测定实验前, 将冷冻后的血浆置于37 ℃的水浴中解冻。血浆以4 000 r·min-1离心5 min, 收集上清液。根据需要将其pH值调整到7.4±0.1。将2 μL的待测前药化合物或阳性对照物溶液(100 μmol·L-1) 加入至98 μL空白血浆中, 37 ℃水浴孵育。在每个时间点(0、10、30、60和120 min) 分别加入400 μL的反应中止液(200 ng·mL-1甲糖宁和200 ng·mL-1拉贝洛尔乙腈溶液) 并充分混合以沉淀蛋白, 并以4 000 r·min-1离心15 min。每份取上清液50 μL与100 μL超纯水混合, 800 r·min-1离心10 min, 然后进行LC-MS/MS分析。
5 药代动力学测定动物实验通过上海药明康德新药开发有限公司的实验动物伦理委员会批准, 动物福利和实验过程遵循AAALAC规定(实验室动物饲养管理和使用标准)。取6只健康雄性SD大鼠(200±10 g), 随机分为2组, 分别口服给予25 mg·kg-1的剂量的前药化合物9f和9g。于不同时间点(0、0.25、0.5、1.0、2.0、4.0、8.0和12 h) 采集全血样品, 分离血浆, 经有机溶剂处理后采用LC-MS/MS法, 分别测定了每组大鼠血浆中氨曲南的浓度。使用Phoenix WinNonlin 6.3软件按照非房室模型计算药代动力学参数。除样品分析期间, 血浆样品收集后置于-20 ℃冰箱保存。
致谢: 目标化合物的核磁共振氢谱、碳谱以及高分辨质谱由中国医学科学院医药生物技术研究所分析测试中心测定, 血浆酯酶代谢稳定性和体内药代动力学评价委托上海药明康德新药开发有限公司测定。
作者贡献: 郭志浩负责所有目标物的合成、文献的调研整理及初稿的撰写; 张昕彤和陈阳负责化合物细胞毒性的测定; 卢曦负责化合物抗菌活性的测定; 宋丹青对实验过程中出现的问题及论文撰写提供了有益的指导; 李迎红负责实验把关、稿件修改等工作并提供了必要的资金支持。以上作者均已通读手稿并同意论文的刊发。
利益冲突: 所有作者均声明不存在利益冲突。
| [1] |
Xu P. Rational clinical application of aztreonam[J]. World Latest Med Inf (世界最新医学信息文摘), 2018, 18: 140, 143. |
| [2] |
Ramsey C, MacGowan AP. A review of the pharmacokinetics and pharmacodynamics of aztreonam[J]. J Antimicrob Chemother, 2016, 71: 2704-2712. DOI:10.1093/jac/dkw231 |
| [3] |
Xu ZQ. Rational design of prodrugs and application[J]. Sci Technol Vision (科技视界), 2018, 22: 203-204. |
| [4] |
Jubeh B, Breijyeh Z, Karaman R. Antibacterial prodrugs to overcome bacterial resistance[J]. Molecules, 2020, 25: 1-16. |
| [5] |
Zhou XL, Wang RX. Application of prodrug design in cephalosporins[J]. World Notes Antibiot (国外医药抗生素分册), 2004, 25: 244-248. |
| [6] |
Gordon EM, Duncton MAJ, Gallop MA. Orally absorbed derivatives of the β-lactamase inhibitor avibactam. design of novel prodrugs of sulfate containing drugs[J]. J Med Chem, 2018, 61: 10340-10344. DOI:10.1021/acs.jmedchem.8b01389 |
| [7] |
Gordon EM, Duncton MAJ, Wang BJ, et al. Toward orally absorbed prodrugs of the antibiotic aztreonam. Design of novel prodrugs of sulfate containing drugs. Part 2[J]. ACS Med Chem Lett, 2020, 11: 162-165. DOI:10.1021/acsmedchemlett.9b00534 |
| [8] |
Beaumont K, Webster R, Gardner I, et al. Design of ester prodrugs to enhance oral absorption of poorly permeable compounds: challenges to the discovery scientist[J]. Curr Drug Metab, 2003, 4: 461-485. DOI:10.2174/1389200033489253 |
| [9] |
Bahar FG, Ohura K, Ogihara T, et al. Species difference of esterase expression and hydrolase activity in plasma[J]. J Pharm Sci, 2012, 101: 3979-3988. DOI:10.1002/jps.23258 |
| [10] |
Swabb EA, Sugerman AA, Stern M. Oral bioavailability of the monobactam aztreonam (SQ 26, 776) in healthy subjects[J]. Antimicrob Agents Chemother, 1983, 23: 548-550. DOI:10.1128/AAC.23.4.548 |