 2021, Vol. 56
2021, Vol. 56



2. 中国科学院大学, 北京 100049
2. University of Chinese Academy of Sciences, Beijing 100049, China
几十年前, 药物研究者在新药研发的早期阶段将重点放在化合物的生物活性上, 而对药物代谢动力学性质的优化与投入有限, 导致很多体外高活性的候选药物因药物代谢动力学性质较差而终止研发。药代动力学主要问题包括首过效应、口服生物利用度较低、易代谢失活以及产生毒性代谢产物等。20世纪80年代后期, 药物研究人员逐渐认识到化合物的理化性质会对其药代动力学性质产生重要影响, 因此相继建立了多种体外代谢实验模型, 如肝微粒体和肝细胞稳定性实验等, 大大降低了因药代动力学性质不佳引起的药物研发损耗[1]。一个理想的候选药物, 不仅应具有高效低毒的特点, 还应具有良好的生物利用度和理想的半衰期, 使其能够在靶器官达到足够的浓度发挥疗效, 而不产生蓄积毒性。本课题组前期分别从提高药物代谢稳定性与血浆稳定性、改善水溶性、提高血脑屏障通透性、降低潜在毒性等方面, 对先导化合物结构优化策略进行了系列综述[2-6]。除此之外, 清除率也是一个十分重要的药代参数——首过代谢清除直接影响到口服药物的生物利用度; 清除率与分布容积共同影响药物的体内半衰期, 进一步影响给药间隔[7]。本文介绍了清除率的概念和研究意义, 以及体内清除率的常见预测方法。重点综述了改善清除率的先导化合物结构优化策略, 主要包括: 通过降低亲脂性、封闭代谢位点、骨架修饰、增加位阻等方法降低肝脏代谢转化清除率; 通过提高亲脂性、降低极性表面积、生物电子等排等方法降低胆汁排泄清除率或肾脏排泄清除率; 最后总结了药物分子的立体构型对清除率的影响。通过这些先导化合物的结构优化策略, 能够显著改善先导化合物的清除率, 为新药设计与研发提供理论指导和实践经验。
1 清除率的概念和研究意义 1.1 清除率的概念清除率(clearance, CL) 定义为: 单位时间内, 由消除器官完全有效地清除含药液体的体积, 其单位通常为mL·(min·kg)-1或L·(h·kg)-1[7]。清除率主要用于反映药物分子在体循环中被提取和消除的快慢程度, 其在药物的研发和临床应用中是一项重要的考察指标。清除率的计算方法如公式(1) 所示(dose: 给药剂量; AUC0-∞: 血药浓度-时间曲线下面积)。
| $ {\rm{CL=dose/AU}}{{\rm{C}}_{{\rm{0 - }}\infty }} $ | (1) |
清除率的大小与给药途径密切相关, 通常考察经静脉注射(i.v.) 给药后药物分子的清除率, 由于这种情况下所有药物分子均达到体循环, 因此计算所得的清除率又称为系统清除率(CLs)。
而若采用口服(p.o.) 给药, 则计算所得清除率称为口服清除率(CLpo), 有时也称为表观清除率(CLapp), 其与系统清除率之间的关系如公式(2) 所示(F: 口服生物利用度)。此外, 在考察药物代谢动力学性质的过程中, 以血液为分析对象, 测得的清除率称为血液清除率(CLb); 以血浆为分析对象测得的清除率称为血浆清除率(CLp)。
| $ {\rm{C}}{{\rm{L}}_{{\rm{po}}}}={\rm{ C}}{{\rm{L}}_{\rm{s}}}/F$ | (2) |
清除率是药物化学工作者进行结构改造过程中需要考虑的最重要的药代参数之一, 因为它决定了药物分子的半衰期、口服生物利用度等其他药代参数[7]。同时, 在确定合适的给药剂量时, 也需要考虑药物分子的清除率[8]。
清除率与分布容积(Vd) 共同决定了药物的消除速率(kel), 并最终影响到药物的半衰期(t1/2)。公式3和4展示了在一房室模型中, 通过分布容积与清除率估算半衰期的方法:
| $ {\rm{CL}} \approx {V_{\rm{d}}} \times {k_{{\rm{el}}}} $ | (3) |
| ${t_{1/2}} \approx {\rm{ln }}2{\rm{ }} \times {V_{\rm{d}}}/{\rm{CL}}$ | (4) |
大多数药物分子通过口服给药, 药物在胃肠道吸收不完全或首过代谢会降低药物的生物利用度。对于主要经肝脏清除的药物, 肝脏提取是影响口服生物利用度和清除率的重要因素。三者之间的关系如公式5和6所示(Fa: 吸收分数; Fg: 药物逃逸肠壁代谢的比例; Eh: 肝脏提取率; Q: 肝脏血流)。
| $F={F_{\rm{a}}} \times {F_{\rm{g}}} \times {\rm{ }}(1 - {E_{\rm{h}}})$ | (5) |
| $ {E_{\rm{h}}}={\rm{ CL}}/Q $ | (6) |
另一方面, 清除率是联系稳态平均血药浓度(Css, av) 和给药剂量的重要纽带(公式7, dose: 给药剂量; τ: 给药间隔)。另外, 生物利用度也受到清除率的影响, 因此在公式7中, 清除率同时影响分子与分母, 即清除率的调节会对系列化合物的给药剂量产生直接影响。
| $ {\rm{dose }}={C_{{\rm{ss, av}}}} \times {\rm{ CL }} \times \tau /F $ | (7) |
通过以上描述可知, 清除率过高的药物, 体内半衰期短, 口服生物利用度差, 血药浓度难以维持在起效浓度以上, 导致药物体内药效不佳; 而清除率过低的药物, 易在体内蓄积产生毒性, 对于安全窗较小的药物尤其需要关注其血药浓度的变化。通常以肝全血流量作为参考评价清除率。清除率大于70%肝全血流量的药物为高清除率药物, 清除率小于30%肝全血流量的药物为低清除率药物。部分动物及人的体重、肝全血流量及清除率范围如表 1所示[9]。
| Table 1 Body weight, hepatic blood flow and clearance range of some animals and human |
肝脏和肾脏是药物清除的两大器官。药物清除的机制主要分为三大类: 代谢转化、胆汁排泄和肾脏排泄(图 1)。此外, 在肠道、皮肤等器官中也可能发生药物清除。

|
Figure 1 Major mechanisms of drug clearance |
肝脏是药物的主要代谢器官, 肝细胞中含有大量的摄取/外排转运体以及丰富的药物代谢酶, 如: Ⅰ相代谢酶CYP450酶、环氧化物水解酶、醛脱氢酶等; Ⅱ相代谢酶葡萄糖醛酸转移酶、谷胱甘肽巯基转移酶等[2, 10]; 有机阴离子多肽转运体(OATP)、有机阳离子转运体(OCT)、乳腺癌耐药蛋白(BCRP)、多药耐药相关蛋白(MRP) 等转运体[11, 12]。药物分子通过被动扩散或主动摄取进入肝细胞后, 一方面, 在不同代谢酶的催化下发生Ⅰ相代谢和(或) Ⅱ相代谢被清除; 也有一些药物不经代谢转化, 直接以原型药物的形式通过胆汁排泄清除。药物及其代谢物的胆汁排泄通常为转运体介导的主动运输过程, 且存在较大的种属差异。
肾脏是药物及其代谢物的主要排泄器官。肾脏排泄主要包括三个过程, 即肾小球滤过、肾小管主动分泌和肾小管重吸收。未与血浆蛋白结合的游离小分子药物都会经肾小球滤过作用进入近端肾小管(公式8, CLr: 药物的肾脏清除率; fu, b: 血液中游离药物分数; GFR: 肾小球滤过率)。脂溶性较高的分子, 容易在肾小管重吸收回到血液循环, 这类分子通常经Ⅰ相和(或) Ⅱ相代谢转化后形成极性代谢物而被排泄。
| ${\rm{C}}{{\rm{L}}_{\rm{r}}}={f_{{\rm{u, b}}}} \times {\rm{ GFR}}$ | (8) |
在改善药物清除率的过程中, 应首先明确药物分子的主要清除途径。药物的清除途径主要受分子的理化性质与透膜性的影响(表 2)[8, 11]。亲脂性高的药物分子往往透膜性好, 易在肾小管重吸收, 因此不易经肾脏排泄, 主要经肝脏代谢转化清除。极性大、透膜性差的药物, 主要经胆汁或肾脏排泄清除, 其跨膜运输受转运体影响较大, 清除途径较复杂[13]。
| Table 2 Influence of physicochemical properties and permeability on drug clearance path |
已知的体内清除率的预测方法有近30种, 主要分为两大类: 体外体内外推法(in vitro-in vivo extrapolation, IVIVE) 和异速放大法(allometric scaling, AS)。
2.1 体外体内外推法体外体内外推法是最常用的一种肝脏清除率预测方法。采用IVIVE法预测药物的肝脏清除率主要经历以下三个步骤: ①测量药物分子在体外肝代谢模型中的内在清除率(CLint, in vitro); ②通过基于生理学的放大参数(PBSF) 换算得到药物分子的肝脏内在清除率(CLint, in vivo); ③选择合适的肝脏处置模型预测药物分子在体内的肝脏清除率(CLh)[14]。
肝微粒体和肝细胞是最实用的两种体外肝代谢模型, 对于主要经CYP450酶代谢清除的分子, 通过将化合物与已知浓度的肝微粒体或肝细胞共孵育, 测得化合物的消除速率(kel), 即可根据表 3中的公式计算出该化合物的体外内在清除率(CLint, in vitro)。随后, 将CLint, in vitro换算为CLint, in vivo, 这一步骤本质上是将实验中使用的蛋白或细胞浓度换算为肝脏内蛋白或细胞的总含量。以小鼠肝微粒体为例, 若将某一小分子与小鼠肝微粒体共孵育, 测得该小分子的消除速率kel, 则根据实验中肝微粒体浓度可计算得到该小分子在小鼠肝微粒体中的内在清除率CLint, in vitro。CLint, in vitro乘以每克肝脏中肝微粒体的含量(即表 4中的换算系数47), 再乘以肝脏质量在小鼠总体重中的占比(54.9), 即可得到小鼠肝脏对该化合物的内在清除率(CLint, in vivo) 的预测值。
| Table 3 Prediction of drug clearance through in vitro-in vivo extrapolation. aCprotein: Concentration of liver microsomes; Ccell: Concentration of hepatocytes; PBSF: Physiological based scaling factors, see Table 4 |
| Table 4 Physiological based scaling factors[9] |
最后, 需要选择合适的肝脏处置模型预测化合物在体内的肝脏清除率(CLh)。图 2中列出了3种最常用的肝脏处置模型以及在相应的模型中, 肝脏清除率(CLh) 的计算方法[14]。其中, 充分搅拌模型是最简单的一种数学模型。在该模型中, 肝脏对药物的清除率由肝脏血流量(Q)、肝脏对药物的内在非结合清除率(CLint, in vivo) 以及药物分子的游离分数(fu) 共同决定, 即将上述计算所得CLint, in vivo代入公式9, 即可得到化合物在体内的肝脏清除率的预测值。
| $ {\rm{C}}{{\rm{L}}_{\rm{h}}}={\rm{ }}(Q \times {f_{\rm{u}}} \times {\rm{ C}}{{\rm{L}}_{{\rm{int}}, in\;vivo}}){\rm{ }}/{\rm{ }}(Q+{f_{\rm{u}}} \times {\rm{ C}}{{\rm{L}}_{{\rm{int}}, in\;vivo}}) $ | (9) |

|
Figure 2 Hepatic disposition models[14]. Cin: Influx drug concentration; Ch: Hepatic drug concentration; Cout: Efflux drug concentration; CLh: Hepatic clearance; Qh: Hepatic blood flow |
采取体外肝代谢模型对体内清除率进行预测会存在一些误差, 例如: 化合物渗透性较差时, 预测值偏高; 化合物经非CYP酶代谢时, 预测值偏低; 若化合物为摄取转运体底物, 则预测值偏低, 因为体外测试中使用的肝细胞与体内肝细胞相比, 细胞膜上转运体活性较低。
2.2 异速放大法异速放大法是一种经验性方法, 假设药物在不同动物间的药代特征是类似的, 差异仅在体重[15]。具体来说, 测量同一药物分子在不同种属动物体内的清除率(CL), 其与动物体重(BW) 之间存在幂函数关系(公式10)[8]。若将公式10等号左右同时取对数, 即可得到线性函数, 即公式11。公式11中的Log a即为图 3中的截距, b为斜率。将人的体重带入公式11, 即可推测出该药物分子在人体内的清除率。在异速放大法的实际应用中, 并不需要测量出药物分子在所有实验动物体内的清除率, 有时只需要测量化合物在某一种或两种动物体内的清除率, 即可根据相应的公式推测出其在人体内的清除率。基于异速放大理论的清除率预测方法有很多种, 表 5中列出了准确性较高的3种[15-17], 通过将清除率预测值与测量值对比, 约60%的化合物, 其清除率测量值与预测值之间的误差在2倍之内; 约80%的化合物, 其清除率测量值与预测值之间的误差在3倍之内; 而100%的化合物清除率测量值与预测值之间的误差在10倍之内。采用异速放大法预测人体内清除率的误差主要来源于: ①药物主要经肝脏代谢, 且在肝微粒体中的稳定性存在较大的种属差异; ②药物分布过程涉及转运体; ③个别种属存在代谢饱和现象等。
| $ {\rm{CL=aB}}{{\rm{W}}^{\rm{b}}} $ | (10) |
| $ {\rm{Log}}\;{\rm{CL=Log}}\;{\rm{a + bLog}}\;{\rm{BW}} $ | (11) |

|
Figure 3 Basics of an allometric scaling experiment[8] |
| Table 5 Three allometric scaling-based prediction methods of human clearance with relatively high accuracy |
通过先导化合物结构优化改善清除率的策略主要包括: 采用降低亲脂性、封闭代谢位点、骨架修饰、增加位阻等方法降低肝脏代谢转化清除率; 采用提高亲脂性、降低极性表面积、生物电子等排等方法降低胆汁排泄清除率或肾脏排泄清除率。此外, 药物的立体构型也会对其清除率产生影响。
3.1 降低药物肝脏代谢转化清除率亲脂性高的药物分子, 主要经肝脏代谢转化而清除。通过化学结构修饰提升化合物的代谢稳定性, 可有效降低肝脏代谢转化清除率, 优化药物分子的药代动力学特征。降低肝脏代谢转化清除率的结构改造策略有: 降低亲脂性、封闭代谢位点、骨架修饰、增加空间位阻降低谷胱甘肽反应性等。
3.1.1 降低亲脂性通常亲脂性高、透膜性好的化合物, 容易进入肝脏发生代谢转化而被清除。体内的大多数代谢酶都具有与亲脂性基团相结合的活性口袋, 对于具有相同骨架或相似结构的化合物, 其清除率往往与亲脂性正相关。因此, 降低亲脂性是改善化合物清除率的重要策略之一。
非洛地平(felodipine, 1) 是第二代钙离子通道阻滞剂(图 4, IC50=0.2 nmol·L-1)[18], 由于其具有较高的亲脂性(cLog P=5.30), 非洛地平在犬体内的系统清除率较高[CL = 38 mL·(min·kg)-1], 血浆半衰期仅为1 h。通过去除苯环3位氯原子, 并在二氢吡啶环的侧链引入极性的氨基侧链, 得到第三代长效型钙离子通道阻滞剂氨氯地平(图 4, amlodipine, 2, IC50=7.9 nmol·L-1)[19]。氨氯地平的亲脂性相比非洛地平降低约2个Log单位, 犬体内系统清除率降低至11 mL·(min·kg)-1, 仅为非洛地平的1/4, 血浆半衰期延长至30 h。氨氯地平的氨基侧链具有碱性, 在生理条件下携带正电荷, 可与携带负电荷的细胞膜进行结合, 这一分子结构特征也决定了其对血管平滑肌细胞钙离子通道具有持久的阻滞作用。

|
Figure 4 Reducing hepatic metabolic clearance of calcium channel blockers by reducing lipophilicity |
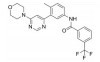
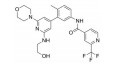
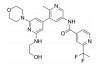
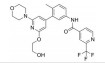
化合物3对C-Raf原癌基因丝苏氨酸蛋白激酶(CRAF) 具有较强的抑制活性(表 6, CRAF IC50=0.000 1 μmol·L-1), 但其对RAS突变的人退行性肺癌细胞Calu-6的增殖(cell proliferation, CP) 抑制活性较低(CP Calu-6 EC50=5.28 μmol·L-1), 且人肝微粒体内的清除率较高[CLint, HLM=43.3 μL·(min·mg)-1][20]。通过在其嘧啶环的2位引入氨基乙醇侧链得到化合物4, 其侧链氧原子与靶标蛋白RAF激酶的595位苯丙氨酸残基形成较强的氢键相互作用, 其细胞增殖抑制活性增强(CP Calu-6 EC50=0.16 μmol·L-1), 相比3提高了近33倍, 但由于化合物4具有较高的亲脂性(cLog P=4.1), 人肝微粒体清除率升高[CLint, HLM=48.0 μL·(min·mg)-1]。将化合物4的嘧啶环与苯环均由吡啶环替换, 得到化合物5, 其亲脂性相比化合物4降低了0.9个Log单位, Chemdraw软件预测结果表明, 右侧苯环采用吡啶环替换对其亲脂性的降低具有较大贡献。化合物5在人肝微粒体中的清除率下降至20.5 μL·(min·mg)-1, 约为化合物4的1/2。进一步将化合物5的三氟甲基中的一个氟原子用甲基替代, 得到化合物6, 亲脂性相比3降低1.1个Log单位, 人肝微粒体中的清除率进一步下降为化合物5的1/2 [CLint, HLM=10.4 μL·(min·mg)-1], 随着化合物亲脂性减弱, 清除率降低(表 6)。进一步改造过程中, 将氨基乙醇侧链替换为乙二醇侧链, 得到细胞增殖抑制活性较高(CP Calu-6 EC50=0.47 μmol·L-1), 且清除率较低[CLint, HLM=13.5 μL·(min·mg]-1) 的化合物7 (LXH254)。值得注意的是, 化合物7的亲脂性高于化合物5, 但其人肝微粒体清除率却相比化合物5有所降低, 这说明亲脂性并不是影响药物清除率的唯一因素, 例如若分子中含有易被药物代谢酶识别的基团, 则可能造成化合物亲脂性降低而清除率升高的特例。另一方面, cLog P只是一个预测值, 可能存在误差, 化合物5与化合物7的cLog P只相差0.3, 实际上两者的亲脂性可能是相似的, 甚至化合物5的亲脂性更高。LXH254在小鼠、大鼠及比格犬体内的系统清除率分别为19、31和3.5 mL·(min·kg)-1。目前, LXH254已进入临床Ⅱ期试验, 用于结肠癌、非小细胞肺癌、卵巢癌等多种癌症的治疗。
| Table 6 Reducing hepatic metabolic clearance of CRAF inhibitors by reducing lipophilicity |

化合物8是美国Epizyme公司通过高通量筛选发现的蛋白精氨酸甲基转移酶(PRMT5) 抑制剂, 该化合物对PRMT5具有一定抑制活性(表 7, PRMT5 IC50=326 nmol·L-1)[21]。通过结构优化得到化合物9, 虽然其对PRMT5的抑制活性明显提升(PRMT5 IC50=13 nmol·L-1), 但体外清除率较高, 经IVIVE法预测所得体内清除率接近人或小鼠肝脏血流[HLM scaled CL=17 mL·(min·kg)-1; MLM scaled CL=80 mL·(min·kg)-1]。通过将吡啶环替换为四氢吡喃环得到化合物10, 其亲脂性降低1.39个Log单位, 清除率也随之降低[HLM scaled CL=10 mL·(min·kg)-1; MLM scaled CL=61 mL·(min·kg)-1], 进一步将四氢吡喃环替换为氧杂环丁烷, 得到化合物11 (EPZ015666), 其亲脂性相比化合物10降低约1个Log单位, 清除率进一步降低[HLM scaled CL < 5 mL·(min·kg)-1; MLM scaled CL=20 mL·(min·kg)-1]。EPZ015666现作为体内工具药用于PRMT5相关药理机制研究[22], 其在小鼠、大鼠和比格犬体内的系统清除率分别为30.0、36.8和46.5 mL·(min·kg)-1[23]。
| Table 7 Reducing hepatic metabolic clearance of PRMT5 inhibitors by reducing lipophilicity. aHLM scaled CL: In vivo intrinsic clearance determined from in vitro intrinsic clearance of human liver microsomes. bMLM scaled CL: In vivo intrinsic clearance determined from in vitro intrinsic clearance of mouse liver microsomes |
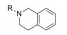
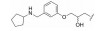
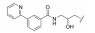
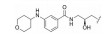
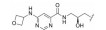
对于代谢途径较明确的化合物, 可以通过封闭代谢位点的结构修饰策略降低化合物的代谢转化清除率, 改善药代动力学特性。
化合物12具有一定的抗锥虫病活性(表 8, T. cruzi IC50=0.63 μmol·L-1), 其在人肝微粒体中的清除率为9.57 mL·(min·kg)-1[24]。分析表明, 在人肝微粒体中, 化合物12主要发生5, 6位双羟基化代谢。将6位氯原子替换为给电子的乙基, 得到化合物13, 其抗锥虫活性提高(T. cruzi IC50=0.14 μmol·L-1), 但在人肝微粒体中的清除率略微上升[CLint, HLM=10.4 μL·(min·mg)-1]。在化合物苯环6位引入硝基或三氟甲基等吸电子取代基, 化合物的抗锥虫活性和人肝微粒体清除率均得到改善[14, T. cruzi IC50=0.04 μmol·L-1, CLint, HLM=1.57 μL·(min·mg)-1; 15, T. cruzi IC50=0.1 μmol·L-1, CLint, HLM=0.94 μL·(min·mg)-1]。考虑到硝基可能在还原的过程中产生自由基引发毒性, 所以选择化合物15进行进一步改造。通过在化合物15的苯环5位引入氟原子封闭羟基化代谢位点, 得到化合物16, 其抗锥虫活性进一步提高(T. cruzi IC50=0.079 μmol·L-1), 化合物16在人肝微粒体中的清除率显著下降[CLint, HLM=0.41 μL·(min·mg)-1], 仅为化合物12人肝微粒体中清除率的1/23。
| Table 8 Reducing hepatic metabolic clearance of anti-T. cruzi agents by blocking the hydroxylation metabolic sites |
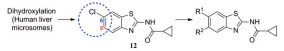
化合物17是一种二氢叶酸还原酶(DHFR) 抑制剂, 其对金葡菌的最小抑菌浓度(MIC) 为0.156 μg·mL-1, 但它在小鼠肝微粒体中的清除率高达173 mL·(min·kg)-1[25]。体外分析表明, 化合物17的主要代谢途径为苯环上甲氧基的O-去甲基代谢(图 5)。采用氘代甲氧基(18) 或三氟甲氧基(19) 替换甲氧基(表 9), 可有效减少O-去烷基化代谢, 降低化合物的清除率[18, CLint, MLM=154 μL·(min·mg)-1; 19, CLint, MLM=22 μL·(min·mg)-1]。采用氯原子替代甲氧基, 封闭O-去甲基代谢位点, 得到化合物20, 其抗菌活性和小鼠肝微粒体中清除率得到较好的平衡[MIC=0.312 μg·mL-1; CLint, MLM=25 μL·(min·mg)-1]。化合物20为消旋体, 其活性单体21 (S构型) 在小鼠肝微粒体中的清除率仅为化合物17小鼠肝微粒体清除率的1/10。经腹腔注射给药后, 化合物21在小鼠体内的清除率为5.96 mL·(min·kg)-1。

|
Figure 5 The proposed metabolic scheme for compound 17 following incubation with mouse liver microsomes |
| Table 9 Reducing hepatic metabolic clearance of DHFR inhibitors by blocking the O-demethylation metabolic site |
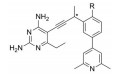
化合物22是经高通量筛选得到的具有亚型选择性的α-氨基-3-羟基-5-甲基-4-异噁唑丙酸(AMPA) 受体拮抗剂, 其在表达跨膜AMPA受体调控蛋白γ-8 (TARP γ-8) 的细胞中表现出一定的AMPA受体拮抗活性(IC50=2.97 μmol·L-1)[26]。但化合物22在人及大鼠肝微粒体中的消除率较高, 分别为81.0%和84.0%, 并且在大鼠体内的清除率[CLrat=234 mL·(min·kg)-1] 超过肝脏血流, 提示除肝脏代谢清除外, 还存在其他清除途径。考虑化合物22结构中的酚羟基可能是关键的Ⅱ相代谢位点。将苯酚基团替换为吲唑(23、24) 或苯并噻唑酮(25、26), 可有效封闭Ⅱ相代谢位点, 降低化合物在大鼠体内的清除率(表 10), 但化合物的肝微粒体消除率依然较高。
| Table 10 Reducing hepatic metabolic clearance of AMPAR antagonists by blocking the phase Ⅱ metabolic site |

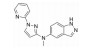
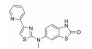
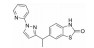
体外研究表明, 化合物26经肝细胞孵育后的主要代谢途径为吡啶环上的氧化代谢(图 6)[26]。其中, 最易发生氧化代谢的位点为吡唑环的对位, 在该位点引入氟原子(27) 或极性侧链(28~30), 可有效封闭氧化代谢位点, 降低化合物在人和大鼠肝微粒体中的消除率。其中, 化合物30 (LY3130481) 的AMPA受体拮抗活性较优(表 11, AMPA IC50=0.065 μmol·L-1), 且在人和大鼠肝微粒体中的消除率均较低, 分别为16.4%和32.3%。化合物LY3130481在大鼠体内的口服生物利用度高达97%, 暴露量为4 220 μg·h·L-1, 经静脉注射给药后的清除率为11.4 mL·(min·kg)-1, 仅为化合物22在大鼠体内系统清除率的1/20。体内研究表明, LY3130481经口服给药后表现出良好的抗惊厥效应, 且不会对大鼠的运动能力造成损伤。

|
Figure 6 Predicted sites of CYP2D6-mediated oxidative metabolism of compound 20 using the StarDrop model. Sites are labeled with a predicted percentage of products formed due to metabolism at that position |
| Table 11 Reducing hepatic metabolic clearance of AMPAR antagonists by blocking the oxidative metabolic site |
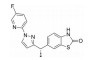
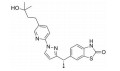
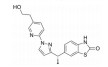
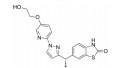
骨架修饰也是降低化合物代谢转化清除率的重要结构改造策略之一。通过骨架修饰可以改变化合物的母核, 调节化合物的理化性质。采用代谢稳定的环系结构替代不稳定的结构片段, 可有效降低化合物的代谢转化清除率。
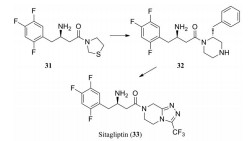
化合物31是默克公司研发的DPP-Ⅳ抑制剂(DPP-Ⅳ IC50=119 nmol·L-1), 其体内药代动力学性质较差, 在大鼠体内的清除率高达120 mL·(min·kg)-1, 生物利用度仅为3% (表 12)[27]。将噻唑烷替换为哌嗪环, 并在哌嗪环上引入苄基, 得到化合物32, 其DPP-Ⅳ抑制活性提高5倍(DPP-Ⅳ IC50=19 nmol·L-1), 但体内清除率仍较高[28]。对其进行代谢产物分析, 发现该化合物的主要代谢途径为哌嗪环上的氧化代谢。在后续的改造中, 研究人员采用代谢稳定的三唑并哌嗪环替换哌嗪环, 并在三唑环上引入三氟甲基, 最终得到DPP-Ⅳ抑制活性高(DPP-Ⅳ IC50=18 nmol·L-1), 且药代性质良好的化合物西格列汀(sitagliptin)[29]。西格列汀在大鼠及犬体内的生物利用度分别为76%和100%, 显著优于化合物31。其在大鼠体内的清除率为化合物31的1/2 [CLrat=60 mL·(min·kg)-1], 并且在犬体内的清除率较低[CLdog=6.0 mL·(min·kg)-1], 半衰期达4.9 h。2006年, 西格列汀作为首个口服DPP-Ⅳ抑制剂上市用于2型糖尿病的治疗。
| Table 12 Reducing hepatic metabolic clearance of DPP-Ⅳ inhibitors by scaffold modification |
趋化因子受体4 (CXCR4) 拮抗剂34具有较高的CXCR4拮抗活性(CXCR4 IC50=6.5 nmol·L-1), 但其碱性较强(pKa=10.7), 透膜性差, 在人及小鼠肝微粒体中保留百分比分别为77.0%和17.0%[30, 31]。并且34对CYP2D6具有一定的抑制活性(CYP2D6 IC50=0.3 μmol·L-1), 提示其可能会在联合治疗中产生药物-药物相互作用。通过去除碱性的丁胺侧链, 并在四氢异喹啉环上引入哌嗪环得到化合物35。35的哌嗪环上的NH与原丁胺侧链NH2取向相似, 保持了CXCR4拮抗活性(表 13, CXCR4 IC50=6.08 nmol·L-1), 同时保护四氢异喹啉结构不被CYP450酶识别, 在人及小鼠肝微粒体中的保留百分比分别提高到99.7%和19.9%。但35的透膜性仍较差, 通过在哌嗪环上不同位置引入甲基(36~38), 虽然可在一定程度上改善化合物的透膜性, 但始终无法在活性、透膜性和清除率之间达到平衡。最终在四氢异喹啉的苯环上引入哌嗪并四氢吡咯的双环结构, 得到化合物39和40, 其中(R)-构型单体40的CXCR4拮抗活性及透膜性均较好(CXCR4 IC50=3.59 μmol·L-1, Pc=740 nm·s-1), 并且在人及小鼠肝微粒体中的保留百分比较适中, 分别为52.2%和36.7%。
| Table 13 Reducing hepatic metabolic clearance of CXCR4 antagonists by scaffold modification |
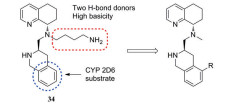
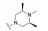
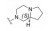
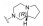
化合物41是第一代苯基噻唑类抗菌药, 对耐甲氧西林金葡菌(MRSA) 的最小抑菌浓度为4.8 μg·mL-1 (图 7)[32]。但由于该化合物含有Schiff氏碱的结构, 易水解代谢失活, 在人肝微粒体中的清除率高达80.3 μL·(min·mg)-1。通过将亚胺并入嘧啶环, 得到化合物42, 其在人肝微粒体中的清除率降低为化合物41的1/24 [CLint, HLM=3.3 μL·(min·mg)-1], 对MRSA的抑菌活性提高, 其MIC为3.12 μg·mL-1。将化合物42的嘧啶环上的氨基替换为肼基, 得到化合物43, 其对MRSA的抑菌活性相比化合物41提高12倍(MIC=0.40 μg·mL-1), 清除率降低至3.0 μL·(min·mg)-1。且与41相比, 化合物43的人肝微粒体半衰期由28.8 min延长至308 min, 提高约10倍。单剂量(50 mg·kg-1) 口服给药后, 43在大鼠体内的半衰期长达8.24 h, 暴露量为100 010.96 μg·h·L -1。

|
Figure 7 Reducing hepatic metabolic clearance of phenylthiazole antibiotics using scaffold hopping strategy |
化合物44 (LY2881835) 是礼来公司研发的GPR40激动剂, 其对人源GPR40招募β-arrestin的EC50为8.7 nmol·L-1 (图 8)[33], 曾进入Ⅰ期临床用于治疗2型糖尿病。临床Ⅰ期数据表明其在人体内的清除率较高(CL=183 mL·min-1)。将化合物中的苄基linker用三唑并吡啶取代, 规避了O-/N-去烷基代谢, 减弱了化合物的两性离子特征, 显著改善了化合物的水溶性和清除率[34]。经异速放大预测, 化合物45 (LY3104607) 在人体内的清除率约为16.7 mL·min-1, 仅为化合物44的1/11。

|
Figure 8 Reducing hepatic metabolic clearance of GPR40 agonists by scaffold modification. HLM: Human liver microsomes; MLM: Mouse liver microsomes; RLM: Rat liver microsomes; DLM: Dog liver microsomes. aSolubility refers to the grams of compounds could dissolve in every 100 g water |
一些药物分子通过与靶标蛋白发生共价结合产生药理活性, 这类分子往往含有迈克尔受体的结构特征, 因此可在谷胱甘肽巯基转移酶(GST) 的催化下与谷胱甘肽(GSH) 结合而被清除。通过增加位阻降低化合物与GST的亲和性, 可以有效降低化合物的清除率。
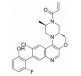
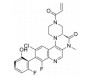
化合物46对KRASG12C的IC50为0.211 μmol·L-1 (表 14), 它的主要清除机制为丙烯酰胺基团发生谷胱甘肽结合[35]。在其哌嗪环上引入甲基得到化合物47, 由于哌嗪环上的乙醚基和甲基均采取直立键的取向, 空间位阻限制了GSH从两侧接近丙烯酰胺, GSH反应半衰期延长(GSH t1/2=416 min)。但甲基的引入也使得化合物的亲脂性增加(Log D=3.8), 人肝微粒体清除率提高[CLint, HLM=32 μL·(min·mg) -1]。将吗啉环替换成内酰胺, 得到化合物48, 其亲脂性相比化合物47降低0.9个Log单位, 人肝微粒体清除率降低为21 μL·(min·mg)-1, 同时GSH反应半衰期延长至585 min。最后在化合物48的哌嗪环上引入甲基得到49, 其人肝微粒体清除率保持在较低水平[CLint, HLM=21 μL·(min·mg)-1], GSH反应半衰期达到929 min, 相比化合物46延长近2倍。
| Table 14 Reducing glutathione reactivity of KRAS inhibitors by increasing steric hindrance |
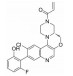
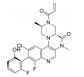
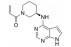
化合物50是一个选择性的JAK3共价抑制剂(IC50=56 nmol·L-1)[36]。体外研究表明, 化合物50在大鼠肝微粒体内的清除率小于15 μL·(min·mg) -1, 但其在大鼠血液中的半衰期仅为98 min, 这是因为其在大鼠体内的清除主要由谷胱甘肽巯基转移酶(GST) 而不是CYP450所介导。在丙烯酰胺的烯基末端引入甲基, 增加位阻, 得到化合物51 (表 15), 其与JAK3的亲和性显著降低, JAK3抑制活性丧失(JAK3 IC50 > 10 000 nmol·L-1)。而在距离丙烯酰胺较远的哌嗪环上引入甲基, 得到化合物52 (PF-06651600), 其GSH反应性降低, 在人血中的半衰期超过360 min, 在大鼠血液中的半衰期达到161 min。这一改造通过稳定化合物与靶标蛋白之间的共价相互作用, 提高了化合物的JAK3抑制活性(JAK3 IC50=33 nmol·L-1)。PF-06651600在人体内清除率的预测值为5.6 mL·(min·kg)-1[37]。
| Table 15 Reducing glutathione reactivity of JAK3 inhibitors by increasing remote steric hindrance |
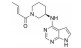
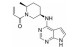
一些极性大的药物分子, 通常是不能在肠内被重吸收的有机阴离子或阳离子, 可以经转运体(P-gp、BCRP、MRP2等) 介导从肝细胞内分泌进入胆汁。对于这类药物分子, 可以通过提高亲脂性、生物电子等排等结构优化策略降低胆汁排泄清除率。
3.2.1 提高亲脂性Log D是与胆汁排泄相关的最重要的理化性质参数之一。对于一些主要经胆汁排泄的药物分子, 可以通过提高亲脂性, 降低极性表面积(PSA) 的方法改善清除率。
选择性电压门控钠离子通道NaV1.7抑制剂53 (hNaV1.7 IC50=13 nmol·L-1) 在人、大鼠及犬肝微粒体中的清除率均较低[表 16, CLint, HLM=14 μL·(min·mg)-1; CLint, RLM=23 μL·(min·mg)-1; CLint, DLM < 5 μL·(min·mg)-1], 但在大鼠和犬体内的清除率却远高于IVIVE预测值[CLrat=58.0 mL·(min·kg)-1, CLdog=10.0 mL·(min·kg)-1], 表明该化合物存在非代谢清除, 即胆汁排泄[38]。采用亲脂性的氯原子替代极性基团氰基, 得到化合物54, 其亲脂性略有提高(Log D=0.8), 极性表面积降低(TPSA=107 Å2), 体内清除率有所降低[CLrat=40.3 mL·(min·kg)-1, CLdog=2.0 mL·(min·kg)-1]。进一步将哒嗪环替换为酸性更弱的氨基吡唑环, 得到化合物55 (PF-05089771), 其亲脂性相比化合物53提高1.6个Log单位, 在大鼠及犬体内的清除率分别降低至化合物53的1/10和1/4 [CLrat=6.0 mL·(min·kg)-1, CLdog=2.7 mL·(min·kg)-1]。
| Table 16 Reducing biliary excretion clearance of NaV1.7 inhibitors by increasing lipophilicity |
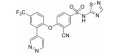
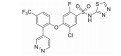

化合物56也是一个选择性NaV1.7抑制剂(NaV1.7 IC50=0.15 μmol·L-1), 其在人及大鼠肝微粒体中的清除率较低, 但在大鼠体内的清除率较高[表 17, CLrat=28.3 mL·(min·kg)-1], 可能与转运体介导的胆汁排泄清除相关[39]。化合物56在犬体内清除率较低, 这也侧面说明了转运体介导的清除存在较大的种属差异。在化合物56的C环上引入亲脂性的氯原子, 并将A环替换为异噁唑, 得到化合物57, 其亲脂性增加(cLog D=2.6), 清除率降低[CLrat=0.78 mL·(min·kg)-1; CLdog=0.05 mL·(min·kg)-1]。由于化合物57具有一定的CYP3A4诱导活性, 经后续改造得到安全性更高的化合物58, 其亲脂性相比化合物56提高0.8个Log单位, 在大鼠及犬体内的清除率较为适中[CLrat=3.83 mL·(min·kg)-1; CLdog=0.38 mL·(min·kg)-1], 活性也得到了进一步提高(NaV1.7 IC50=0.036 μmol·L-1)。经口服给药后, 化合物58在C57Bl/6小鼠体内展现出良好的抗组胺效果。
| Table 17 Reducing biliary excretion clearance of NaV1.7 inhibitors by increasing lipophilicity |
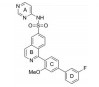
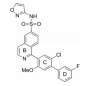

生物电子等排是先导化合物结构优化过程中常用的策略, 采用生物电子等排策略不仅可以提高化合物的药理活性和安全性[40], 也可改善化合物的药代动力学性质。化合物59对P2Y12受体具有较高的亲和力(表 18, IC50=6.3 nmol·L-1), 采用透光率比浊法(LTA) 测定其对二磷酸腺苷(ADP) 诱导的血小板聚积具有一定的抑制作用(LTA IC50=21 nmol·L-1)[41]。其在大鼠体内表现出中等的清除率[CLrat=24 mL·(min·kg)-1], 推测这一现象可能是因为羧基的存在, 导致部分原型药物经胆汁排泄造成的。采用生物电子等排策略, 以磷酸基团替换羧基, 得到若干低清除率的化合物[表 18, 60, CLrat=3.7 mL·(min·kg)-1; 61, CLrat=2.1 mL·(min·kg)-1; 62, CLrat=5.4 mL·(min·kg)-1]。其中, 化合物61在大鼠体内的清除率最低, 且其P2Y12受体亲和力与抗凝血能力均较优(binding IC50=1.0 nmol·L-1, LTA IC50=8.0 nmol·L-1), 因此, 对其进行体内研究, 经静脉注射给药后, 化合物61在大鼠三氯化铁血栓模型中表现出良好的治疗效果。
| Table 18 Reducing biliary excretion clearance of P2Y12 receptor antagonists using bioisosterism strategy |


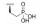
PD-1/PD-L1信号通路与肿瘤免疫逃逸机制密切相关, 目前, 有较多抑制PD-1/PD-L1相互作用的小分子被报道用于抗肿瘤免疫治疗。多数小分子内含有羧基的结构, 例如图 9中的化合物63 (IC50=0.180 nmol·L-1) 和64 (IC50=112 nmol·L-1)[42, 43]。这些小分子在体内清除较快, 化合物63经口服给药(10 mg·kg-1) 后, 在小鼠体内的半衰期仅1.6 h。化合物64经静脉注射给药(4 mg·kg-1) 后, 在大鼠体内的半衰期约1.38 h, 清除率高达74.5 mL·(min·kg)-1, 超过大鼠肝脏血流。虽然对这些小分子抑制剂药代性质的研究还不是很深入, 但可以推测这些携带羧基的小分子可能作为转运体底物被富集于肝脏, 进而被代谢或经胆汁排泄清除[12]。若采用生物电子等排策略将羧基替换为其他基团, 或许可降低其体内清除率。

|
Figure 9 Inhibitors of PD-1/PD-L1 |
一些大极性、水溶性较好的分子易经肾脏排泄。但对于肾功能损伤的患者来说, 这类药物的清除率相比健康人会有所降低, 从而引起体内蓄积毒性, 并进一步增加患者肾脏的负担。对于这一类药物分子, 应通过增加化合物的亲脂性, 减少肾脏排泄, 增加非肾清除率。
去氨加压素(desmopressin) 是抗利尿激素的类似物(图 10), 对抗利尿激素受体(V2R) 具有很强的激动活性(hV2R EC50=0.2 nmol·L-1), 在多个国家批准上市用于治疗糖尿病尿崩症[44]。去氨加压素在人体内的药代动力学研究表明, 其在健康人体内的系统清除率约1.7 mL·(min·kg)-1, 半衰期为2.8 h; 而在肾功能损伤者体内的系统清除率降低至0.69 mL·(min·kg)-1, 相应半衰期延长至10 h。去氨加压素在大鼠体内的清除率[7.5 mL·(min·kg)-1] 接近肾小球滤过率, 非肾清除率仅占11%。将其4位的谷氨酰胺残基替换为亲脂性更高的缬氨酸残基, 得到活性和选择性更高的化合物66 (图 10, hV2R EC50=0.05 nmol·L-1), 其亲脂性增加, 在HPLC中的容量因子k'由3.72增加到4.56, 在大鼠体内的清除率也有所提高[CLrat=9.6 mL·(min·kg)-1]。对化合物66的2、3、4位氨基酸残基进行修饰得到活性与选择性较高的化合物, 并以此为基础, 对二硫键及7位脯氨酸残基进行修饰, 得到化合物67~71, 随着化合物亲脂性的增加(即HPLC中的容量因子k'增加), 化合物在大鼠体内非肾清除率占比增加(表 19)。其中, 化合物70在大鼠体内的系统清除率最高, 为42 mL·(min·kg)-1, 相比去氨加压素提高5倍, 且非肾清除率占比提高至60%。

|
Figure 10 Reducing renal excretion clearance by increasing lipophilicity |
| Table 19 Reducing renal excretion clearance of hV2R agonists by increasing lipophilicity |
药物分子中含有手性中心时, 应分别对消旋体及不同构型单体的代谢性质进行考察。一方面, 不同构型的单体与代谢酶之间的亲和力可能不同, 导致两者清除率差异较大。另一方面, 若两种构型单体被同一个代谢酶代谢转化, 则以消旋体形式给药时, 对映异构体之间竞争同一代谢酶的结合位点, 导致两者的清除率与以单体形式给药时不同。因此, 当化合物具有手性中心时, 应结合消旋体与单体的活性与药代性质, 选择合适的给药形式。
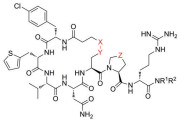
阻转异构体是一种特殊的手性异构体: 由于围绕单键旋转受阻导致构象异构体之间无法相互转换, 则这些构象异构体互为阻转异构体。PI3Kβ抑制剂72 (IC50=3 nmol·L-1) 是一对阻转异构体, 其中, 优势构型为P构型(PI3Kβ IC50=2 nmol·L-1)[45]。两种构型的单体在人肝微粒体中孵育后都表现出较低的清除率, 但两者在人肝细胞中的清除率相差了20多倍[表 20, (M)-72, hHep CLint=32.8 mL·(min·kg)-1, (P)-72, hHep CLint=1.33 mL·(min·kg)-1]。这是由于(M)-72是醛氧化酶(AO) 的底物, 因此在肝细胞中清除较快, 而(P)-72不被AO识别。(P)-72在不同实验动物体内均表现出低至中等的清除率[大鼠: 11.3 mL·(min·kg)-1; 比格犬: 3.67 mL·(min·kg)-1; 食蟹猴: 1.83 mL·(min·kg)-1; 恒河猴: 1.67 mL·(min·kg)-1]。
| Table 20 Influence of axial chirality on activity and hepatic metabolic clearance of PI3Kβ inhibitors. aHLM CLint: In vivo intrinsic clearance determined from human liver microsomes fortified with NADPH and UDGPA. bhHep CLint: In vivo intrinsic clearance determined from cryopreserved human hepatocytes |
普罗帕酮是Ic类抗心律失常药物, 其(S)-型异构体的β受体拮抗作用是(R)-型异构体的100倍(表 21)[46]。两种构型单体的代谢均主要由CYP2D6介导。若以单一构型给药, (S)-型异构体的清除率(CLpo=2521 mL·min-1) 大于(R)-型异构体(CLpo=1 279 mL·min-1), 且因达不到有效浓度, 无法产生β受体阻断作用[47]。但若长期以消旋体给药, 由于(R)-型异构体对CYP2D6的亲和性强于(S)-型异构体, 自身代谢清除率提高(CLpo=1 460 mL·min-1), (S)-型异构体清除率下降(CLpo=920 mL·min-1), (S)-型异构体血药浓度反而高于(R)-型异构体。因此, 综合考虑活性与清除率, 普罗帕酮以消旋体形式给药可以达到最理想的治疗效果。
| Table 21 Influence of spatial configuration on the activity and pharmacokinetic properties of propafenone |
药物的手性构型也会对肾脏排泄产生影响。肾小管上皮细胞的转运体对手性药物的不同构型可能具有不同的选择性, 且由于转运体具有饱和性, 不同异构体之间可能会竞争转运蛋白的结合位点。例如, 在消旋的氧氟沙星中, (R)-型异构体会抑制肾小管对(S)-型异构体的分泌(表 22)[48]。因此, 若以消旋体给药, 可观察到(S)-氧氟沙星的肾脏清除率[CLrenal=7.14 L·(h·1.73 m2)-1] 低于(R)-氧氟沙星[CLrenal=7.53 L·(h·1.73 m2)-1]。由于氧氟沙星和左氧氟沙星的抗菌谱和适应症略有不同, 两者在临床上都有使用。
| Table 22 Pharmacokinetic parameters of (S)-(-)-ofloxacin and (R)-(+)-ofloxacin after oral administration of (±)-ofloxacin (200 mg) to healthy volunteers. a1.73 m2: Human standard body surface area |
在先导化合物结构优化的早期阶段, 重视对化合物药代性质的考察可以显著降低研发失败率, 减少损耗。清除率作为联系口服生物利用度、半衰期、稳态血药浓度等多个药代参数的纽带, 是药物化学工作者需要着重考察的指标。降低药物清除率可提高药物的口服生物利用度、延长药物分子的体内半衰期; 对于清除率过低的药物, 也应采取合适的策略提高其体内清除率, 以防产生蓄积毒性。随着对药物清除机制及其影响因素的深入了解, 越来越多的体外代谢评价模型和体内清除率预测方法得到开发。在对化合物进行结构修饰时, 应采用适当的清除率评价体系预测药物分子的体内清除率与主要清除机制, 通过合理的结构优化改善清除率, 使药物达到安全性、活性和药代动力学性质的最佳平衡, 这样才能有效提高药物研发的成功率。
作者贡献: 张蕊负责调研文献, 整理撰写文章; 王江负责审阅、修改文章; 朱浩然负责调研文献, 核对数据; 柳红负责审阅、修改文章。
利益冲突: 本文不存在任何与本稿件相关的利益冲突。
| [1] |
Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates?[J]. Nat Rev Drug Discov, 2004, 3: 711-715. DOI:10.1038/nrd1470 |
| [2] |
Wang J, Liu H. Lead compound optimization strategy (1) - changing metabolic pathways and optimizing metabolism stability[J]. Acta Pharm Sin (药学学报), 2013, 48: 1521-1531. |
| [3] |
Liu H, Wang J, Lin D, et al. Lead compound optimization strategy (2) - structure optimization strategy for reducing toxicity risks in drug design[J]. Acta Pharm Sin (药学学报), 2014, 49: 1-15. |
| [4] |
Li Z, Wang J, Zhou Y, et al. Lead compound optimization strategy (3) - structure modification strategies for improving water solubility[J]. Acta Pharm Sin (药学学报), 2014, 49: 1238-1247. |
| [5] |
Hong Y, Zhou Y, Wang J, et al. Lead compound optimization strategy (4) - improving blood-brain barrier permeability through structural modification[J]. Acta Pharm Sin (药学学报), 2014, 49: 789-799. |
| [6] |
Wu X, Wang J, Liu H. Lead compound optimization strategy (6) - improving the plasma stability[J]. Acta Pharm Sin (药学学报), 2018, 53: 192-201. |
| [7] |
Hutzler JM, Ring BJ, Anderson SR. Low-turnover drug molecules: a current challenge for drug metabolism scientists[J]. Drug Metab Dispos, 2015, 43: 1917-1928. DOI:10.1124/dmd.115.066431 |
| [8] |
Smith DA, Beaumont K, Maurer TS, et al. Clearance in drug design[J]. J Med Chem, 2019, 62: 2245-2255. DOI:10.1021/acs.jmedchem.8b01263 |
| [9] |
Ring BJ, Chien JY, Adkison KK, et al. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 3: comparative assessement of prediction methods of human clearance[J]. J Pharm Sci, 2011, 100: 4090-4110. DOI:10.1002/jps.22552 |
| [10] |
Fan PW, Zhang D, Halladay JS, et al. Going beyond common drug metabolizing enzymes: case studies of biotransformation involving aldehyde oxidase, γ-glutamyl transpeptidase, cathepsin b, flavin-containing monooxygenase, and ADP-ribosyltransferase[J]. Drug Metab Dispos, 2016, 44: 1253-1261. DOI:10.1124/dmd.116.070169 |
| [11] |
El-Kattan AF, Varma MVS. Navigating transporter sciences in pharmacokinetics characterization using the extended clearance classification system[J]. Drug Metab Dispos, 2018, 46: 729-739. DOI:10.1124/dmd.117.080044 |
| [12] |
Song X, Wang J, Liu H. Lead compound optimization strategy (8) - drug transporters and related drug design strategies[J]. Acta Pharm Sin (药学学报), 2021, 56: 432-444. |
| [13] |
Varma MV, Steyn SJ, Allerton C, et al. Predicting clearance mechanism in drug discovery: extended clearance classification system (ECCS)[J]. Pharm Res, 2015, 32: 3785-3802. DOI:10.1007/s11095-015-1749-4 |
| [14] |
Sodhi JK, Benet LZ. Successful and unsuccessful prediction of human hepatic clearance for lead optimization[J]. J Med Chem, 2021, 64: 3546-3559. DOI:10.1021/acs.jmedchem.0c01930 |
| [15] |
Tang H, Hussain A, Leal M, et al. Interspecies prediction of human drug clearance based on scaling data from one or two animal species[J]. Drug Metab Dispos, 2007, 35: 1886-1893. DOI:10.1124/dmd.107.016188 |
| [16] |
Tang H, Mayersohn M. A novel model for prediction of human drug clearance by allometric scaling[J]. Drug Metab Dispos, 2005, 33: 1297-1303. DOI:10.1124/dmd.105.004143 |
| [17] |
Wajima T, Fukumura K, Yano Y, et al. Prediction of human clearance from animal data and molecular structural parameters using multivariate regression analysis[J]. J Pharm Sci, 2002, 91: 2489-2499. DOI:10.1002/jps.10242 |
| [18] |
Leonardi A, Motta G, Pennini R, et al. Asymmetric N-(3, 3-diphenylpropyl)aminoalkyl esters of 4-aryl-2, 6-dimethyl-1, 4-dihydropyridine-3, 5-dicarboxylic acids with antihypertensive activity[J]. Eur J Med Chem, 1998, 33: 399-420. DOI:10.1016/S0223-5234(98)80015-9 |
| [19] |
Arrowsmith JE, Campbell SF, Cross PE, et al. Long-acting dihydropyridine calcium antagonists. 1. 2-Alkoxymethyl derivatives incorporating basic substituents[J]. J Med Chem, 1986, 29: 1696-1702. DOI:10.1021/jm00159a022 |
| [20] |
Ramurthy S, Taft BR, Aversa RJ, et al. Design and discovery of N-(3-(2-(2-hydroxyethoxy)-6-morpholinopyridin-4-yl)-4-methylphenyl)-2-(trifluorom ethyl)isonicotinamide, a selective, efficacious, and well-tolerated RAF inhibitor targeting RAS mutant cancers: the path to the clinic[J]. J Med Chem, 2020, 63: 2013-2027. DOI:10.1021/acs.jmedchem.9b00161 |
| [21] |
Duncan KW, Rioux N, Boriack-Sjodin PA, et al. Structure and property guided design in the identification of PRMT5 tool compound EPZ015666[J]. ACS Med Chem Lett, 2016, 7: 162-166. DOI:10.1021/acsmedchemlett.5b00380 |
| [22] |
Chan-Penebre E, Kuplast KG, Majer CR, et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models[J]. Nat Chem Biol, 2015, 11: 432-437. DOI:10.1038/nchembio.1810 |
| [23] |
Rioux N, Duncan KW, Lantz RJ, et al. Species differences in metabolism of EPZ015666, an oxetane-containing protein arginine methyltransferase-5 (PRMT5) inhibitor[J]. Xenobiotica, 2016, 46: 268-277. DOI:10.3109/00498254.2015.1072253 |
| [24] |
Fleau C, Padilla A, Miguel-Siles J, et al. Chagas disease drug discovery: multiparametric lead optimization against trypanosoma cruzi in acylaminobenzothiazole series[J]. J Med Chem, 2019, 62: 10362-10375. DOI:10.1021/acs.jmedchem.9b01429 |
| [25] |
Lombardo MN, G-Dayanandan N, Keshipeddy S, et al. Structure-guided in vitro to in vivo pharmacokinetic optimization of propargyl-linked antifolates[J]. Drug Metab Dispos, 2019, 47: 995-1003. DOI:10.1124/dmd.119.086504 |
| [26] |
Gardinier KM, Gernert DL, Porter WJ, et al. Discovery of the first alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonist dependent upon transmembrane ampa receptor regulatory protein (TARP) γ-8[J]. J Med Chem, 2016, 59: 4753-4768. DOI:10.1021/acs.jmedchem.6b00125 |
| [27] |
Xu J, Ok HO, Gonzalez EJ, et al. Discovery of potent and selective beta-homophenylalanine based dipeptidyl peptidase Ⅳ inhibitors[J]. Bioorg Med Chem Lett, 2004, 14: 4759-4762. DOI:10.1016/j.bmcl.2004.06.099 |
| [28] |
Brockunier LL, He J, Colwell LF, et al. Substituted piperazines as novel dipeptidyl peptidase Ⅳ inhibitors[J]. Bioorg Med Chem Lett, 2004, 14: 4763-4766. DOI:10.1016/j.bmcl.2004.06.065 |
| [29] |
Kim D, Wang L, Beconi M, et al. (2R)-4-Oxo-4-[3-(trifluoromethyl)-5, 6-dihydro[1, 2, 4]triazolo[4, 3-a]pyrazin-7(8H)-yl]-1-(2, 4, 5-trifluorophenyl)butan-2-amine: a potent, orally active dipeptidyl peptidase Ⅳ inhibitor for the treatment of type 2 diabetes[J]. J Med Chem, 2005, 48: 141-151. DOI:10.1021/jm0493156 |
| [30] |
Nguyen HH, Kim MB, Wilson RJ, et al. Design, synthesis, and pharmacological evaluation of second-generation tetrahydroisoquinoline-based CXCR4 antagonists with favorable ADME properties[J]. J Med Chem, 2018, 61: 7168-7188. DOI:10.1021/acs.jmedchem.8b00450 |
| [31] |
Truax VM, Zhao H, Katzman BM, et al. Discovery of tetrahydroisoquinoline-based CXCR4 antagonists[J]. ACS Med Chem Lett, 2013, 4: 1025-1030. DOI:10.1021/ml400183q |
| [32] |
Seleem MA, Disouky AM, Mohammad H, et al. Second-generation phenylthiazole antibiotics with enhanced pharmacokinetic properties[J]. J Med Chem, 2016, 59: 4900-4912. DOI:10.1021/acs.jmedchem.6b00233 |
| [33] |
Hamdouchi C, Kahl SD, Patel Lewis A, et al. The discovery, preclinical, and early clinical development of potent and selective GPR40 agonists for the treatment of type 2 diabetes mellitus (LY2881835, LY2922083, and LY2922470)[J]. J Med Chem, 2016, 59: 10891-10916. DOI:10.1021/acs.jmedchem.6b00892 |
| [34] |
Hamdouchi C, Maiti P, Warshawsky AM, et al. Discovery of LY3104607: a potent and selective G protein-coupled receptor 40 (GPR40) agonist with optimized pharmacokinetic properties to support once daily oral treatment in patients with type 2 diabetes mellitus[J]. J Med Chem, 2018, 61: 934-945. DOI:10.1021/acs.jmedchem.7b01411 |
| [35] |
Kettle JG, Bagal SK, Bickerton S, et al. Structure-based design and pharmacokinetic optimization of covalent allosteric inhibitors of the mutant GTPase KRASG12C[J]. J Med Chem, 2020, 63: 4468-4483. DOI:10.1021/acs.jmedchem.9b01720 |
| [36] |
Thorarensen A, Dowty ME, Banker ME, et al. Design of a Janus kinase 3 (JAK3) specific inhibitor 1-((2S, 5R)-5-((7H-pyrrolo[2, 3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one (PF-06651600) allowing for the interrogation of JAK3 signaling in humans[J]. J Med Chem, 2017, 60: 1971-1993. DOI:10.1021/acs.jmedchem.6b01694 |
| [37] |
Telliez JB, Dowty ME, Wang L, et al. Discovery of a JAK3-selective inhibitor: functional differentiation of JAK3-selective inhibition over pan-JAK or JAK1-selective inhibition[J]. ACS Chem Biol, 2016, 11: 3442-3451. DOI:10.1021/acschembio.6b00677 |
| [38] |
Swain NA, Batchelor D, Beaudoin S, et al. Discovery of clinical candidate 4-[2-(5-amino-1H-pyrazol-4-yl)-4-chlorophenoxy]-5-chloro-2-fluoro-N-1, 3-thiazol-4-ylbenzenesulfonamide (PF-05089771): design and optimization of diaryl ether aryl sulfonamides as selective inhibitors of NaV1.7[J]. J Med Chem, 2017, 60: 7029-7042. DOI:10.1021/acs.jmedchem.7b00598 |
| [39] |
Weiss MM, Dineen TA, Marx IE, et al. Sulfonamides as selective NaV1.7 Inhibitors: optimizing potency and pharmacokinetics while mitigating metabolic liabilities[J]. J Med Chem, 2017, 60: 5969-5989. DOI:10.1021/acs.jmedchem.6b01851 |
| [40] |
Lima LM, Barreiro EJ. Bioisosterism: a useful strategy for molecular modification and drug design[J]. Curr Med Chem, 2005, 12: 23-49. DOI:10.2174/0929867053363540 |
| [41] |
Caroff E, Hubler F, Meyer E, et al. 4-((R)-2-{[6-((S)-3-Methoxypyrrolidin-1-yl)-2-phenylpyrimidine-4-carbonyl]amino}-3-phosphonopropionyl)piperazine-1-carboxylic acid butyl ester (ACT-246475) and its prodrug (ACT-281959), a novel P2Y12 receptor antagonist with a wider therapeutic window in the rat than clopidogrel[J]. J Med Chem, 2015, 58: 9133-9153. DOI:10.1021/acs.jmedchem.5b00933 |
| [42] |
Yang F, Zhang Y, Ying H, et al. Biaryl derivative, preparation method thereof and pharmaceutical application thereof: WO, 2019/149183 A1[P]. 2019-08-08.
|
| [43] |
Guo J, Luo L, Wang Z, et al. Design, synthesis, and biological evaluation of linear aliphatic amine-linked triaryl derivatives as potent small-molecule inhibitors of the programmed cell death-1/programmed cell death-ligand 1 interaction with promising antitumor effects in vivo[J]. J Med Chem, 2020, 63: 13825-13850. DOI:10.1021/acs.jmedchem.0c01329 |
| [44] |
Wisniewski K, Qi S, Kraus J, et al. Discovery of potent, selective, and short-acting peptidic V2 receptor agonists[J]. J Med Chem, 2019, 62: 4991-5005. DOI:10.1021/acs.jmedchem.9b00132 |
| [45] |
Chandrasekhar J, Dick R, Van Veldhuizen J, et al. Atropisomerism by design: discovery of a selective and stable phosphoinositide 3-kinase (PI3K) β inhibitor[J]. J Med Chem, 2018, 61: 6858-6868. DOI:10.1021/acs.jmedchem.8b00797 |
| [46] |
Kroemer HK, Funckbrentano C, Silberstein DJ, et al. Stereoselective disposition and pharmacologic activity of propafenone enantiomers[J]. Circulation, 1989, 79: 1068-1076. DOI:10.1161/01.CIR.79.5.1068 |
| [47] |
Kroemer HK, Fromm MF, Buhl K, et al. An enantiomer-enantiomer interaction of (S)-propafenone and (R)-propafenone modifies the effect of racemic drug-therapy[J]. Circulation, 1994, 89: 2396-2400. DOI:10.1161/01.CIR.89.5.2396 |
| [48] |
Okazaki O, Kojima C, Hakusui H, et al. Enantioselective disposition of ofloxacin in humans[J]. Antimicrob Agents Chemother, 1991, 35: 2106-2109. DOI:10.1128/AAC.35.10.2106 |