 2021, Vol. 56
2021, Vol. 56

慢性髓细胞白血病(CML) 和成人急性淋巴白血病(ALL) 的发病原因是血液祖细胞的9号和22号染色体发生交互易位, 导致BCR和ABL1基因在费城染色体(Ph) 上融合, 表达的BCR-ABL1融合蛋白具有ABL1激酶的活性结构域, 使得许多信号通路异常活化, 导致白血病的发生、分化、增殖的失控。已有治疗CML的药物如伊马替尼(1, imatunib)、尼洛替尼(2, nilotinib) 等BCR-ABL1激酶抑制剂都是有效药物, 作用靶标是ATP结合位点, 是结合于DFG-out构象的Ⅱ型抑制剂。达沙替尼(3, dasatinib) 也是作用于ATP结合位点, 但是结合于DFG-in构象的Ⅰ型抑制剂。然而持续的药物治疗都因发生BCR-ABL1T315I的突变而失效, Thr315是激酶的门户氨基酸, 逃逸了1~3的抑制性结合。尽管Ⅱ型的帕那替尼(4, ponatinib) 对发生突变的肿瘤有治疗效果, 也因结合于ATP位点, 对其他激酶发生脱靶而有不良反应。这些替尼的不良反应是因为结合的ATP位点是酪氨酸激酶的共有结构。阿思尼布的研究目标不是BCR-ABL1T315I的ATP位点, 而是作用于“远离”该位点的变构位点。
|
2003年有人发现豆蔻酰基(十四烷酰基) 参与ABL1的自调节过程(Hantschel O, Nagar B, Guettler S, et al. Amyristoyl/phosphotyrosine switch regulate sc-Abl. Cell, 2003, 112: 845-857), 该自调节作用是, 当豆蔻酰基进入ABL1的C端豆蔻酰基结合腔, 与N端发生共价结合, 致使ABL1降低了构象的柔性而成非活化态, 这是个天然的自抑制作用。当形成BCR-ABL1的融合态蛋白时, BCR占据了ABL1的N端帽区的豆蔻酰结合位点, 导致了BCR-ABL1的结构性活化, 致使细胞发生持续性分化增殖而恶性变。BCR-ABL1的构象与激酶ATP位点的DFG-out不同, 与之结合的抑制剂之间不会发生交叉耐药, 这是研制ABL1变构抑制剂的分子依据(Fabbroa D, Manleya PW, Jahnke G, et al. Inhibitors of the Abl kinase directed at either the ATP- or myristate-binding site. Biochim Biophys Acta, 2010, 1804: 454-462)。
2 活性评价用野生型BCR-ABL1wt的ABL1164-515片段和突变型的BCR-ABL1T325I蛋白分别测定化合物对野生型和突变型的抑制活性(IC50), 评价受试物在酶水平上的选择性抑制作用。用BCR-ABL1wt和BCR-ABL1T325I分别转染的小鼠造血细胞Luc-Ba/F3模型评价细胞水平上的选择性抑制作用(IC50)。
3 先导化合物 3.1 苗头化合物寻找苗头化合物的方法是基于片段的分子设计(FBDD), 用NMR信号检测。由于旨在寻找变构位点的抑制剂, 用伊马替尼预先占据ABL1的ATP结合位点, 以确保信号的变化不是结合ATP位点所致。筛选了500个水溶性和结构多样性小分子, 通过滴定法测定ABL1-伊马替尼复合物特定1H谱化学位移的变化, 确定苗头物与ABL1的结合常数, 或者用滴定法测定受试物竞争性的排代工具药GNF-2 (5) 与ABL1复合物的Kd值, 5是已知结合于豆蔻酰基变构域的化合物, Kd = 7.4 μmol·L-1。这两种NMR测定方法有高度重合性。在发现的30个苗头中, 化合物6对ABL1具有一定的亲和力Kd = 6 μmol·L-1, 相对分子质量MW = 185, 计算配体效率较高(LE = 0.6), 因而是良好的苗头。
|
首先变换氯原子为甲基、三氟甲基或氟原子, 都使结合力降低, 而溴置换氯元素结合性提高5倍。羧酸酯和氨基未做变换可能认为是与靶标ABL1结合的必需基团。然而6对BCR-ABL1的生化功能没有影响, 对转染该融合蛋白的Ba/F3细胞即使在50 μmol·L-1浓度下都没有抑制活性, 提示6只有结合没有功能。为了揭示为什么化合物6结合于豆蔻基结合腔而未呈现功能, 研究了化合物6-ABL1-伊马替尼三元复合物的晶体结构, 表明6的结合位置与ABL1的螺旋1形成弯曲的抑制态构象是不相容的。图 1a是三元复合物晶体结构示意图, 伊马替尼和化合物6分别结合于ATP腔和变构区豆蔻基结合腔; 图 1b显示了化合物6处于豆蔻酰基的结合位置, 6的甲酯与Helix 1的Ile521的侧链阻挡了Helix 1转变为非活化构象, 所以没有功能。

|
图 1 a: Crystal structure of ABL1 kinase domain in complex with imatinib and compound 6. b: Structural detail, showing the position of 6 in the myristate pocket of ABL1 kinase |
基于FBDD方法没有(也不可能) 得到有细胞活性的化合物, 但FBDD提供的结构信息经分子模拟可用于指导优化结合强度。根据图 1的信息, 为了模拟豆蔻基的长链将羧基甲酯置换为正己胺基(7), 提高了结合力(Kd = 4 μmol·L-1), 但仍没有细胞活性。模拟工具药5结合于弯曲型Helix1的药效团, 演化为化合物8 (Kd = 6 μmol·L-1), 但仍没有细胞活性。研制者怀疑用1H-NMR是否反映了化合物结合于失活性的弯曲型Helix1构象。因而改用在Helix1上的Val525残基进行15N-标记, 利用15N, 1H-HSQC谱的信号峰的强度判断化合物的结合状态, 结果表明, 直线型的Helix1的Val525信号为尖锐的高峰, 而弯曲状Helix1的Val525只有平均的峰强度。化合物6、7和8的信号是明显的高尖峰, 而工具药5的只为平均的峰强度。从而解释了无功能活性的化合物即便结合于变构区, 并没有影响Helix1的直线型构型, 而5则引起Helix1的弯曲(Jahnke W, Grotzfeld RM, Pelle X, et al. Binding or bending: distinction of allosteric Abl kinase agonists from antagonists by an NMR-based conformational assay. J Am Chem Soc, 2010, 132: 7043-7048)。

|
鉴于化合物5结构左侧的三氟甲氧基是功能活性的必要基团(因为移至其他位置则失去细胞活性), 推测该基团对于稳定Helix1的无活性弯曲构型是重要的药效团, 因而用CF3O-基团替换8的二氯原子, 化合物9对ABL1的结合活性Kd = 10 μmol·L-1, 对野生型BCR-ABL1转染Luk Ba/F3细胞增殖的抑制活性GI50 = 8 μmol·L-1, 这个结果打开了深入研究的道路。进而去除苯环上的酚羟基和氯原子, 并用吗啉环置换N-甲基哌嗪, 化合物10的生化活性和细胞活性都显著提高, Kd = 2 μmol·L-1和GI50 = 2 μmol·L-1。10可视作里程碑化合物。
3.3 结构生物学指导优化结构化合物10与ABL1激酶域和伊马替尼三元复合物晶体结构(图 2) 揭示了以下要点: ① 10结合于豆蔻基结合腔中。CF3O-基处于狭窄结合通道的深部, 氟原子与亮氨酸Leu359的羰基发生静电相互作用。②酰胺基的NH经结构水分子介导与酶中丙氨酸Ala452和谷氨酸Glu481形成氢键网络。③两个芳环都与酶有良好的π-π相互作用。④吗啉环有两种定位: 一是未与酶分子接触, 进入水相; 另一是内翻180°, 进入变构域的疏水裂隙中, 与裂隙的苏氨酸Thr453、甲硫氨酸Met456和脯氨酸Pro480组成的疏水基团发生疏水相互作用, 吗啉环内翻180°的位置提示了进一步设计依据。

|
图 2 Two possible orientations of the morpholine ring of compound 10 in the crystal structure. One mode of morpholine ring points into the solvent phase (green in color); the other flips 180°, binding along a lipophilic cleft at the exit of the myristate channel (yellow in color) |
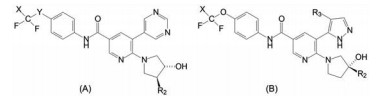
为了利用结合于疏水裂隙的结合空间, 在右侧苯环的3位单键连接嘧啶环, 旨在以平面结构适配于疏水裂隙, 在4位连接有柔性的亲水性基团有利于溶解性。优化的目标定为对ABL1的高抑制活性, 对转染野生型和突变型(T315I) BCR-ABL1的细胞有强抑制活性, 以及适宜的物理化学和体外药理学性质。合成的代表性化合物活性和物化以及药理学性质分别列于表 1和表 2。
| Table 1 Typical compounds and activity against BCR-ABL1 in biochemical assay (IC50) and in cellular proliferation assays (GI50) |
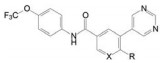

| Table 2 The selective physicochemical and in vitro pharmacological properties. a: Solubility in the fasted state simulated intestinal fluid; b: Rat liver microsome; c: In a radioligand binding (RLB) assay |
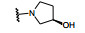
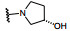
分析构效关系可以看出, 母核为苯环或吡啶环对酶和细胞活性没有显著变化, 但吡啶为母核的化合物log P降低大约1个单位(如化合物11和12), 所以之后的优化固定为吡啶环。吡啶与嘧啶环单键连接, 并且在吡啶4位连接有取代基, 使得两个芳环难以共面, 避免了分子间的π-π叠合增加晶格能, 有利于提高溶解性。4位引入含氮的片段, 如四氢吡咯或哌嗪都有利于降低疏水性, 提高水溶性。其中羟基吡咯的活性和溶解性都占优, 而且S构型18的抑制酶和细胞活性(尤其是发生BCR-ABL1T315I突变的细胞) 强于R构型的17。应该指出的是, 对映体17和18的物化性质应完全相同, 测定的分配系数、pKa和在人工肠液的溶解度的差异应为实验误差所致。虽然18是优化的化合物, 但对HERG蛋白有一定的抑制作用, 因而还得做进一步优化。
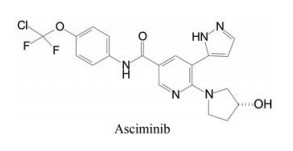
下一步是在保持抑制酶和细胞活性的同时, 消除或降低对HERG钾通道的抑制作用。对HERG通常显示抑制作用的分子多存在含氮的碱性基团, 这与提高溶解性加入碱性基团相矛盾, 在矛盾之下, 仍以提高变构为切入点。高活性化合物18与BCR-ABL1-伊马替尼三元物的晶体结构显示, 左侧苯环的三氟甲氧基的结合部位并没有充满该处空间, 为提高范德华作用, 增大基团体积用两种方式修饰, 一是用硫原子替换氧原子, 另一是将一个氟原子换成氯。18的另一个修饰位点是右侧苯环的4位羟基四氢吡咯环的取代基变换, 以调整母核吡啶环与3位的嘧啶环的扭角(构象), 影响活性和溶解性; 第三个变换位点是对嘧啶环的变换, 基于化合物18的复合物晶体结构发现嘧啶环距离谷氨酸残基Glu481的羰基很近, 有形成氢键的元素, 但需要有氢键给体, 经分子模拟发现3-吡唑基(含有NH而嘧啶没有) 容易生成氢键。综合上述三处的变换, 合成的代表性化合物列于表 3和表 4。从有限的数据分析构效关系如下: ①化合物18的CF3O-基团换成硫代(19) 或氯代(20), 抑制酶或细胞活性都显著提高, 证明充满该处的结合腔是有利的。② 20的羟基四氢吡咯环上再连接一个羟基, 化合物21疏水性(分配系数)进一步降低, 抑酶活性不变, 但细胞活性显著下降, 推测是不利于穿越细胞膜内。③化合物22~25是将嘧啶环换作吡唑, 活性略有下降, 但抑制HERG蛋白作用显著降低, 推测是碱性变弱的缘故。权衡得失, 确定吡唑取代的化合物占优。
| Table 3 Comparison of the inhibition activity for the compounds progressing from pyrimidine (series A) to pyrazole (series B) |
| Table 4 The selective physicochemical and in vitro pharmacological properties. a: The general formula are as same as in Table 3; b: Measured at QPatch automated patch clamp system |
化合物22~25对ABL1激酶和野生型/突变型BCR-ABL1转染的细胞都呈现强效抑制作用(GI50 = 0.6~2.7 nmol·L-1), 而且对HERG蛋白有很弱作用, 进一步研究了对肝微粒体的稳定性和细胞的过膜性(Caco-2) 以及小鼠的药代动力学性质, 这几个化合物都是相近的, 例如口服生物利用度(F) 在53%~100%之间, 清除率(Cl) 为11~26 mL·min-1·kg-1, 半衰期(t1/2) 为0.8~1.1 h。然而将这些化合物灌胃移植肿瘤小鼠, 只有化合物23和25有显著抑制生长活性(数据省略)。最后确定化合物23为候选化合物, 定名为阿思尼布(asciminib), 经临床前和Ⅲ期临床研究, 表明对慢性髓细胞白血病的治疗作用, FDA于2021年2月批准上市。由于阿思尼布结合部位是变构调节剂豆蔻酰基的结合腔, 因而与多数激酶的ATP结合的替尼类药物不发生交叉耐药, 尤其对门户氨基酸残基发生变异的T315I而产生耐药的患者, 阿思尼布是有效的治疗药物(Schoepfer J, Jahnke W, Berellini G, et al. Discovery of asciminib (ABL001), an allosteric inhibitor of the tyrosine kinase activity of BCR-ABL1. J Med Chem, 2018, 61: 8120-8135)。
6 阿思尼布与BCR-ABL1的结合模式阿思尼布与BCR-ABL1和伊马替尼三元复合物晶体结构(图 3a) 提示, 两个药物结合在不同的部位, 分别结合于变构调节域和ATP结合域, 避免了二者发生交叉耐药。图 3b是阿思尼布与变构位点的结合模式, 氯代二氟甲氧基结合于小的疏水腔, 氟原子与Leu359的骨架羰基发生静电作用, 左侧苯环与Thr453和Met456发生疏水作用, 酰胺的羰基氧经结构水与Arg351生成氢键, 吡啶的氮原子经水分子介导形成氢键, 吡唑环结合于疏水裂隙, 并与Ala452羰基发生氢键结合, 所以吡唑处于吡啶的酰基的间位形成U形状是非常必要的。羟基四氢吡咯处于水相边缘, 起到助溶作用。
|

|
Figure 3 a: The bindings of imatinib (red in color) in ATP binding site and asciminib (purple in color) in allosteric domain of BCR-ABL1 protein, respectively. b: The binding mode of asciminib to ABL1 allosteric domain |
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和疗效评价等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多重性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
基于变构域的药物创制(SBDD) 近年来受到药学界的重视, 既往激酶抑制剂的药物研究, 多数集中在ATP结合域为靶标, 一方面是容易发生脱靶效应, 而且靶标一旦发生变异, 导致同类药物失效的局面。干预变构调节的理念可以消除上述忧虑, 因为不同的激酶调节变构的配体不同, 机制和变构域的差异提高了选择性作用, 也避免了交叉耐药和脱靶作用。擅长于基于片段的药物发现(FBDD) 技术的诺华公司从BCR-ABL1融合蛋白的变构机制入手, 通过FBDD发现苗头化合物, 娴熟地运用NMR、X-射线晶体学和分子模拟指导药物设计和构效关系分析, 成功地研制出阿思尼布, 成为以激酶的变构位点为靶标的治疗慢性髓细胞白血病的首创性药物。 (编者按)