 2021, Vol. 56
2021, Vol. 56

20世纪60年代研制非甾类避孕药, 最后研制出治疗乳腺癌的他莫昔芬, 成为首创的选择性雌激素受体调节剂(selective estrogen receptor modulator, SERM), 于1977年FDA批准上市。他莫昔芬称作受体功能调节剂而不是雌激素完全拮抗剂, 是因为存在一部分激动作用, 对幼年大鼠有弱激动作用, 却可完全阻断雌二醇促子宫生长作用。对妇女主要表现抗雌激素活性, 但也有雌激素样作用, 如促进阴道角化和肝脏合成白蛋白等。20世纪90年代业界致力于研制雌受体的完全拮抗剂, 本品即在此背景下展开的。
2 活性评价评价化合物对雌受体的激动和拮抗作用, 是用幼年雌性大鼠子宫发育重量作指标, 通过空白对照、单独给受试物、单独给雌激素, 以及受试物与雌激素联合给药, 评价各个实验组大鼠子宫重量, 并以体重校正(mg子宫重/100 g体重), 按以下公式计算化合物的激动百分率和拮抗百分率:
| $\text { 激动 } \%=\frac{C-A}{B-A} \times 100 \quad \text { 拮抗 } \%=\frac{B-D}{B-A} \times 100 $ |
式中A、B、C和D是经体重校正过的子宫重量, A代表空白对照组、B是单纯雌二醇组、C是单纯受试物, D是雌二醇+受试物组合的子宫重量。
3 先导化合物的设计 3.1 原理雌激素作用方式是与受体结合并诱导受体构象变化, 活化的复合物进入细胞核, 识别了特异的DNA结合位点, 启动转录机制, 影响蛋白质的表达。
设计的化合物希望结合于与雌二醇相同的受体位点, 但同时还用结构中第二个功能基结合于受体结合位点以外的区域, 以改变受体蛋白的构象, 从而阻断雌受体的功能。第二个功能基团应不干扰对雌受体的识别与结合, 为此需经过一定长度的连接基(spacer) 相连。
3.2 7-取代的雌二醇衍生物已有的文献报道, 为了研究用于雌二醇放射性免疫测定的特异性抗体, 研究人员制备了雌二醇的各种衍生物, 发现7位连接直链烷基的化合物保持对雌受体的亲和力(Baulieu EE, Truong H. Parameters influencing the purification of calf uterus estrogen receptor by affinity chromatography. FEBS Lett, 1974, 46: 321-325)。
以此借鉴, 设计的首轮化合物是在雌二醇分子结构的7位经10个亚甲基连接不同功能基的化合物, 结构与活性列于表 1。受试样品为差向异构体混合物, 7α∶7β = 7∶3 (Wakeling AE, Bowler J. Novel antioestrogens without partial agonist activity. J Steroid Biochem, 1988, 31: 645-653)。结果表明, 在C7经饱和链相连的功能基中, 正丁胺的酰胺(5) 呈现完全的拮抗作用, 基本没有激动作用, 而其他化合物都有部分激动作用。进而证明了5的拮抗活性源于α异构体。
| Table 1 The agonistic and antagonistic effects of C7-substituted estradiol derivatives on uterine growth in juvenile rats |
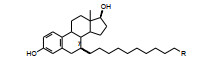
表 1化合物的7位取代基主要是α构型(7R), 进一步的结构优化都是7α取代。由于化合物5拮抗作用最强, 而且没有激动活性, 故以5为新的起点, 对N-丁基作变换, 合成的化合物列于表 2。结果表明, 高拮抗活性的化合物的R为C4~C6 (化合物8和9), 化合物12 (R=新戊基) 也是高活性的拮抗剂, 提示胺基上取代的碳原子数是重要的, 其他化合物为部分拮抗剂。
| Table 2 The effect of 7α substitution on activity |
为了优化7α链的酰基长度合成的化合物列于表 3。结果表明, 从C4到C10, 对雌受体的拮抗作用随碳链加长而提高, 直到纯拮抗剂, 但酰基的碳数还得与胺的烷基碳数相匹配, 例如化合物15的活性显著强于14。7α碳链(酰基+氮上的烷基) 总碳原子数以16~18为宜。
| Table 3 The effect of alkyl chain length on activity |

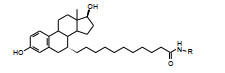
化合物8灌胃给药, 对子宫的生长呈现激动作用, 提示代谢产物与原药的作用差异。8在体内代谢水解成游离酸, 后者有激动活性。为降低代谢作用将酰仲胺置换为酰叔胺, 例如化合物19是8的酰仲胺作N-甲基化, 拮抗作用增强了3倍, 提示酰仲胺上的氢原子没有参与同受体结合作用, 提高了拮抗作用。
因而合成酰叔胺化合物, 结果列于表 4。化合物19皮下注射或灌胃大鼠对受体都有强拮抗作用, 剂量降低为原来的四分之一后仍有高活性。N-乙基化合物20保持有高活性, 进一步加大烷基体积, 则活性下降, 例如N-异丙基化合物22活性显著降低。
| Table 4 The activity of 7α-tertiary amide compounds |

酰仲胺和酰叔胺化合物以不同途径给药, 引起对雌受体激动/拮抗作用的差异, 是由于代谢产物所致, 胺基的氧化脱烷基提高了对雌受体的激动活性, 这是不利的。因而试图将N-烷基支化以增加位阻降低代谢, 合成的化合物列于表 5。构效关系表明带支链的酰仲胺如化合物23和25皮下注射的活性呈现完全拮抗作用, 而相应甲基化产物24和26 (为酰叔胺) 则为部分激动剂, 表明大基团的酰叔胺是不利的因素。
| Table 5 The effect of N-branched alkyl substitution on activity |
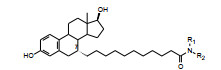
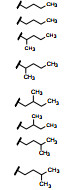
表 6列出的酰仲胺的取代基为苯烷基, 考察刚性环对活性的影响。结果表明, 在保留酰胺氮上氢原子(提供氢键给体) 情况下, 末端苯环会引起化合物的激动活性, 因而不可取。
| Table 6 Activity of secondary amide compounds containing benzene ring at the end |
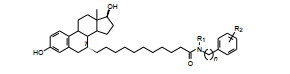
化合物8和19的N-正丁基若被全氟正丁基替换, 34和35仍然保持强抑制活性(表 7), 灌胃大鼠也未呈现雌受体的激动作用, 提示氟代化合物有利于活性和代谢稳定性。
| Table 7 The effect of N-butyl fluoride substitution on activity |
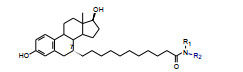
综上所述, 化合物19和35是两个活性最强的化合物, 灌胃大鼠的ED50分别为6和2 mg·kg-1, 提示N-甲基-N-正丁基或N-甲基-N-全氟正丁基等酰叔胺化合物是具有代谢稳定性的完全拮抗剂(Bowler J, Lilley TJ, Pittam JD, et al. Novel steroidal pure antagonists. Steroids, 1989, 54: 71-99; Wakeling AE, Bowler J. Biology and mode of action of pure antiestrogens. J Steroid Biochem, 1988, 30: 141-147)。
5 氟维司群及其作用机制化合物19作为雌受体的完全拮抗剂, 代号为ICI-164384进入临床研究。与此同时对7位侧链作进一步变换。考虑到酰叔胺既没有碱性, 也无氢键形成能力, 推测变换酰胺基团成其他极性基团, 其中包括亚砜基的变换保持拮抗活性, 末端烷基氟代提高了代谢稳定性, 优化出化合物36 (未发表优化过程)。
化合物36代号为ICI-164780, 是雌受体完全拮抗剂, 离体抑制雌受体活性是化合物19的10倍。对幼鼠抑制子宫生长的半数有效剂量(ED50) 为0.06 mg·kg-1, 显著强于19 (ED50 = 0.9 mg·kg-1); 36体外抑制MCF-7人乳腺癌细胞的IC50为0.29 nmol·L-1, 19为1.3 nmol·L-1。36的活性也强于4-羟基他莫昔芬。大鼠或猴一次肌肉注射化合物36, 可持续抑制雌受体活性。对移植性Br10和MCF-7人乳腺癌细胞的裸鼠一次注射化合物36, 效果相当于连续四周每日给药他莫昔芬的效果(Wakeling AE, Dukes M, Bowler J. A potent pure antiestrogen with clinical porential. Cancer Res, 1991, 51: 3867-3873)。
化合物36定名为氟维司群(fluvestrant), 经三期临床研究证明对绝经妇女雌受体呈阳性的乳腺癌是有效的治疗药, 于2002年FDA批准上市。

|
氟维司群最初认为是选择性雌受体调节剂(SERM), 从结构分析, 是在雌二醇的7位连接含有亚砜基的脂肪链, 将雌激素的激动作用翻转为拮抗剂, 以为作用机制与他莫昔芬相似。后来研究发现并不是单纯占据并结合雌受体引起的拮抗作用, 而是结合后导致雌受体的降解失活, 表现为完全拮抗剂。氟维司群与雌受体结合后, 分子中的亲脂性侧链增加了蛋白表面的疏水性, 因之改变了受体蛋白构象, 稳定性降低导致受体蛋白降解(Wu, YL, Yang X, Ren Z, et al. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol Cell, 2005, 18: 413-424)。这样, 氟维司群定为选择性雌受体降解剂(SERD), 其完全拮抗作用的功能即在于此。
氟维司群的口服生物利用度低, 但在体内半衰期很长, t1/2 = 6天, 以油性溶液肌肉注射给药250 mg, 每月一次, 治疗绝经后雌激素受体阳性的局部晚期或转移性乳腺癌。
氟维司群的首创性在于, 它的上市开辟了诱导蛋白降解的小分子药物治疗领域, 其作用犹如酶或催化剂, 与靶标蛋白结合后引起蛋白降解, 本身并没有消耗。
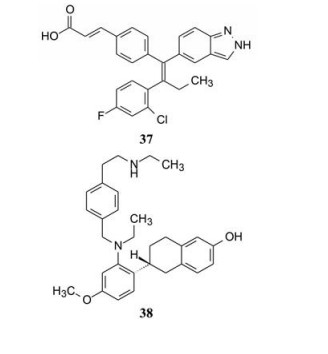
另一个研发的SERD是brilanestrant (37), 终止于II期临床阶段(Lai A, Kahraman M, Govek S, et al. Identification of GDC-0810 (ARN-810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J Med Chem, 2015, 58: 4888-4904)。
|
处于临床研究的另一兼有SERD和SERM双重作用的艾拉司群(38), 低剂量时为SERM, 高剂量为SERD, 因而又称作SERM/SERD杂合剂(SSH), 可能影响受体构象变化的过程与氟维司群有所不同。38还具有组织特异性, 在骨组织中呈现激动作用, 在乳腺和子宫中为拮抗作用, 而且可穿越血脑屏障, 有望治疗乳腺癌转移的脑瘤(Garner F, Shomali M, Paquin D, et al. RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anti-Cancer Drugs, 2015, 26: 948-956)。
氟维司群的成功还引发了研究对其他靶标蛋白的降解剂。例如研发选择性雄受体降解剂(SARD) 治疗前列腺癌(Ponnusamy S, Coss CC, Thiyagarajan T, et al. Novel selective agents for the degredation of androgen receptor variants to treat castration-resistancet tprstate cancer. Cancer Res, 2017, DOI: 10.1158/0008-5472.), 以及由SERD到更广泛的蛋白降解剂, 例如蛋白质水解嵌合体(PROTACs), 对于研发缺少结合位点或结合腔的蛋白靶标的抑制剂(降解剂) 是个重要的平台技术, 意义重大(Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov, 2017, 16: 101-114)。
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和疗效评价等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多重性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
雌受体在不同的组织器官中的分布和作用是不同的, 以他莫昔芬为标志的选择性雌受体调节剂(SERM) 开创了新的治疗领域。多数SERM既有拮抗雌激素作用, 也有部分激动活性。氟维司群是在雌二醇的7位连接亲脂性长链, 在同雌受体结合后, 能使受体呈现不稳定构象以致发生降解。发挥了完全拮抗作用, 因而成为治疗雌受体阳性的乳腺癌长效药物。更重要的是氟维司群作为首创药开辟了选择性雌受体降解剂(SERD) 治疗领域, 并引发了选择性雄激素受体降解剂(SARD) 等药物的研发。 (编者按)