 2021, Vol. 56
2021, Vol. 56

人类肿瘤疾病30%是由于癌基因KRAS突变所引起, 是肺、结肠和胰腺癌等常见的肿瘤驱动基因, 肺癌中20%的KRAS蛋白的Gly12变异成Cys12 (KRASG12C)。在生长因子的信号通路中, KRAS蛋白的功能是个分子开关, 通过在S-IIP区域与非活化型的二磷酸鸟苷(GDP) 结合或转换成与三磷酸鸟苷(GTP) 结合成活化型的开关循环, 调节下游效应蛋白的增殖效应。当KRAS发生G12C突变, 损坏了与GTP酶活化蛋白的结合, 使KRAS失活, 导致细胞的持续增生。KRAS突变在功能上使肿瘤成为永动机, 被称为肿瘤跳动的心脏。
KRAS蛋白突变与癌症密切相关, 已知有三十多年之久了, 但学界和企业界一直没有研制出药物, 原因是由于它的可药性差: 一是KRAS与GDP和GTP酶的结合力非常强, Ki值达pmol·L-1之强度, 外来的抑制剂很难达到与GD(T)P酶发生竞争性结合的强度; 另外, KRAS蛋白的结构除难以竞争GDP结合位点外, 没有其他明显的结合腔穴, 缺乏明显的疏水腔, 缺乏设计抑制剂的抓手。
本品是安进(Amgen) 利用基于片段的药物设计和形成共价的不可逆结合反应, 研制了针对KRAS蛋白变构区隐匿的Cys12的不可逆抑制剂, 将KRAS-GDP复合蛋白锁定在非活性的不可恢复的构象状态。
1.2 既有研发的化合物—二硫化合物及其优化为了找到能结合KRAS的Cys12苗头化合物, Ostrem等采用基于片段分子的方法, 筛选了380个含有二硫键的化合物评价对KRAS-GDP标签性结合, 用质谱检测全蛋白的MS信号, 发现化合物2E07 (1) 和6H05 (2) 具有结合能力。遂以2为起始物进行先导结构的演化(hit-to-lead), 得到了活性较强的化合物3。图 1是3与KRASG12C的共晶结构, 3没有结合在核苷酸结合腔中, 而是在变构区的疏水性沟槽上, 即KRAS-GDP变构区域的S-IIP处, 将KRAS构象固定在非活化状态, 并延伸到Cys12以外的区域中。3的胺基与Gly60羰基形成氢键(Ostrem JM, Peters U, Sos ML, et al. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature, 2013, 503: 548-551)。

|
Figure 1 Co-crystallography of compound 3 binding to KRASG12C |
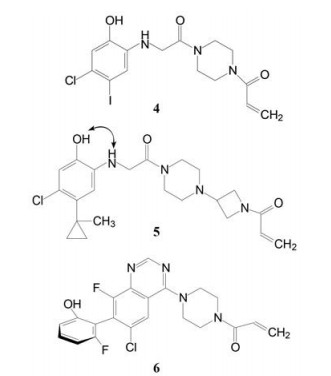
二硫化合物是较弱的亲电性基团, 与巯基发生可逆性共价结合。为提高抑制活性强度, 将二硫结构变换为亲电性稍强的丙烯酰胺和乙烯基磺酰胺, 含有这两种迈克尔反应基团的类似化合物都呈现活性, 由于乙烯基磺酰胺的亲电性强于丙烯酰胺而可能导致选择性差, 因而优化了丙烯酰胺系列。其中化合物4是以哌嗪为连接基, 用LC-MS-MS确证了与KRAS的Cys12发生共价结合, 经生化方法测定了4抑制KRASG12C的速率常数[kobs/[I] = 0.1 (mol·L-1)-1·s-1], 显示出一定的活性。进而优化活性和过膜性, 得到ARS-853 (5), 其活性和成药性都得到了改善, 是KRAS变构抑制剂的里程碑式的工具药。
然而5与KRASG12C共晶结构显示, 哌嗪-氮杂环丁烷-丙烯酰胺链超出了S-IIP的结构域, 而且结构中邻氨基酚和甘氨酸片段都是代谢不稳定的片段, 为此将邻氨基酚变换成喹唑啉骨架, 并且将迈克尔基团经哌嗪环直接连在母核上, 在合成的系列化合物中优化出化合物6。
|
6的喹唑啉8位氟代、7位为2'-OH-6'-F-苯基取代的化合物, 活性显著高于其他在联喹唑啉-苯基的迫位缺少取代基的化合物。6的喹唑啉环与苯环的两面角呈垂直构象, 由于转动受阻导致有阻转异构体的存在, 拆分后S-构型为优映体, 代号为ARS-1620。图 2是ARS-1620与KRASG12C共晶结合图, 分子呈伸展形处于变构区的S-II腔中(Switch-II pocket), 外端的酚羟基经3个结构水分子与Asp69、Arg68和Gln99形成氢键网络, 喹唑啉N1与His95发生氢键结合, 哌嗪旁的羰基与Lys16氢键结合, 迈克尔基团与Cys12发生共价结合(Janes MR, Zhang JC, Li LS, et al. Targeting KRAS mutant cancers with a covalent C12C-specific inhibitor. Cell, 2018, 272: 578-589)。

|
Figure 2 Co-crystallography of compound 6 binding to KRASG12C |
安进公司为了研究KRAS抑制剂, 与Carmot Therapeutics公司合作, 利用Carmet拥有化学型演化(chemotype evolution) 的技术平台, 平台是由含有迈克尔基团的片段分子与有成药骨架的片段分子缩合成单一分子组成的化合物库, 图 3是有代表性的两类片段分子的结构, 左侧是含有丙烯酰胺的片段分子, X为片段偶联的位点。右侧是3 000多个结合左侧亲电性片段的分子。该平台合成出大约两万个结构多样的单个目标分子, 普筛的化合物是未提纯的纳克级样品, 用生化和质谱方法快速评价化合物的结合性能, 达到设定标准的苗头化合物, 制备毫克级的纯净化合物作确证性评价, 从而得到新的苗头化合物7。

|
Figure 3 Chemotype evolution leading to the discovery of 7. Molecules on left hand are fragments where X indicates the inactive handle of the acrylamide. Molecules on the right hand are representative fragments being coupled to target compounds |
图 4是化合物7与KRASG12C共晶的X射线衍射图, 结合要点是丙烯酰胺结合于Cys12, 而且羰基氧与Lys16形成氢键, 另一个羰基与Ala56和Tyr96的羟基形成两组氢键。溴苯基深入到疏水腔中, 溴原子被众多疏水残基环绕。异噁唑酰胺处于Gln99侧链和Switch II之间。

|
Figure 4 Co-crystallography of compound 7 binding to KRASG12C |
将化合物7的酰胺基与溴代苯酚环合成为吲哚以固定母核的构象, 作为探索优化先导物的路径, 合成的代表性化合物列于表 1。结果表明, 变换吲哚3-位连接的末端片段对活性有一定的影响, 但不显著。例如化合物13的四氢异喹啉(THIQ) 没有取代基, 动力学数据[kobs/[I]=308 (mol·L-1)-1·s-1] 和生化转化活性(IC50 =1.27 μmol·L-1) 都较好, 但细胞活性很弱(IC50 =36.5 μmol·L-1)。所以, 变换THIQ片段作为优化部位可能意义不大。综合这三种活性, 化合物9优于其他分子。
| Table 1 Impact of 3-indole substitution on the potency. a: Observed rate (kobs/[I] M-1s-1) derived from 0.02, 0.2, 2, and 20 h of relative % bound at 10 μmol·L-1 compound, according to the equation kobs/[I] = kobs/ln[(100% conversion)/100]/inhibitor concentration/incubation time. b: Dose response GDPKRASG12C/C118A (1-169) SOS1-catalyzed GTP exchange assay. c: Quantification of phospho-ERK1/2 following compound treatment and EGF stimulation by MSD assay in MIA PaCa-2 cells |
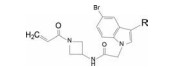
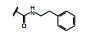

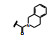
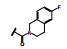
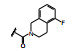


化合物9与KRASG12C的共晶结构如图 5d和5f所示, 这两个复合物都是低能构象, 5d的吲哚环在Arg68侧链的上面, 发生范德华结合, 3-位的酰胺平面与吲哚环的CCCO两面角为155°, 接近于共面性180°。5-甲氧基四氢异喹啉(THIQ) 处于Tyr96和Gln99环之间, 与His95的咪唑环发生π-π叠合。5f的酰胺平面与吲哚环的CCCO两面角为69°, 接近于90°, THIQ所处的位置也与5d不同, 除了与His95的π-π叠合外, 还与Tyr96发生边-面的π-π叠合。两种共晶的其他部位的结合方式相同, 例如与Lys16和Tyr96的氢键结合以及与Cys12的共价键结合等。

|
Figure 5 X-ray cocrystal structure of 9 in complex with KRASG12C |
以化合物9为先导结构, 固定末端的3-甲氧基-THIQ, 变换母核吲哚环上的取代基, 合成的代表性化合物列于表 2。用甲基“扫描”吲哚环的各个位置得到的构效关系如下: ① 2-位甲基(化合物18) 的生化活性强于化合物7一倍, 抑制细胞活性强7倍, 进而2-环丙基取代(19) 活性更强。② 7-甲基取代(20) 的细胞活性没有改善, 但2, 7-二甲基化合物21的细胞活性比7提高38倍。③ 5-氯替换5-溴(22, 23) 活性仍存。特别是2-环丙基-5-氯-7-甲基取代的24的活性更加优良, 抑制细胞活性达到IC50=0.219 μmol·L-1。
| Table 2 Optimization of compound 9 |
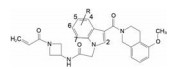
化合物24的较高活性可从复合物单晶X-射线衍射(图 6) 揭示的结合模式加以说明, 概括如下: ① 24的结合模式与9的结合模式大体相同, 但重要的区别是四氢异喹啉环进入到一个本来不存在的“神秘”腔内, 是由于His95的侧链旋转, 形成了由Tyr96、His95和Gln99组成的腔。②吲哚环与酰胺平面的CCCO两面角(64°) 类似于图 5f的结合模式, 2-环丙基的存在降低了扭转两面角的能耗, 同时也增加了范德华作用力, 因而活性提高了。③ 7-甲基处于Thr58、Tyr71和Gly60所在的范德华力结合范围, 这种有利的结合增加了活性。④在所有优化过程中, 发现Switch II (S-II) 是个变化很大的区域, 它的打开和关闭以及非确定性, 还没有找到与共价键结合的相关性(Shin Y, Jeong JW, Wurz RP, et al. Discovery of N-(1-aacryloylazetidin-3-yl)-2-(1H-indol-1-yl)acetamides as a covalent inhibitor of KRASG12C. ACS Med Chem Lett, 2019, 10: 1302-1308)。然而化合物24对啮齿类的口服生物利用度较低, 体内的清除率却很高(数据省略), 故仍需作进一步优化。

|
Figure 6 X-ray cocrystal structure of 24 in complex with KRASG12C |
进一步优化化合物24以改善药代性质和提高体内抗增殖活性似乎难以入手, 因为各片段都是优化而得。为此另辟构建新骨架的路径, 从分析里程碑化合物6和24与KRASG12C的结合模式入手。这两个化合物都是定位于KRASG12C的变构区S-II区域, 如图 7所示, 亲电基团丙烯酰胺都结合于残基Cys12的巯基, 分子的走向和氢键结合也大致相同, 不同的是24的四氢异喹啉进入到S-II域中由Tyr96、His95和Gln99组成的疏水腔中, 而6的氟代苯酚没有进入该疏水腔。

|
Figure 7 Comparison of the GDP-KRASG12C binding modes of 6 and 24. The tetrahydroisoquinoline portion of indole 24 engages a "cryptic pocket" (magenta) not exploited by compound 6, which is induced by side chain rotation of H95 and comprises the residues Tyr96, His95, and Gln99 |
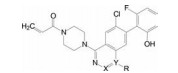
通过叠合图 7中化合物6和24的活性构象, 发现在化合物6的N1位连接出疏水片段相当于24的四氢异喹啉所占的位置, 因而合成了以酞嗪为母核的新类型化合物, 结构和活性列于表 3中。结合共晶结构分析构效关系如下: ①化合物25是在酞嗪环4位连接苯环, 生化测定的交换反应活性和抑制细胞活性都低于化合物6, 分别降低20和70倍。然而25与KRASG12C的共晶结构显示4-苯基进入了Tyr96 His95/Gln99“神秘”腔穴(图 8a)。②苯环的2'-位连接疏水性基团, 如甲基(25)、乙基(28)、异丙基(31) 的生化和细胞活性都依次显著提高。化合物31的活性达到6的水平。③将酞嗪环换成喹唑啉酮环, 相应于2'-异丙基苯的化合物32, 其生化和细胞活性分别提高了3和9倍, 32与KRASG12C的共晶结构显示(图 8b), 苯环上的异丙基与Tyr96发生疏水-疏水相互作用, 是提高活性的重要元件。
| Table 3 Biochemical and cellular activity of new scaffolds designed to access the Tyr96 His95/Gln99 cryptic pocket |

|
Figure 8 X-ray crystallography confirms compound 25 (a) and 32 (b) access the cryptic pocket. Additional van der Waals contacts between the isopropyl moiety of 32 and His95, Tyr96, and Gln99 account for the increased biochemical and cellular potency of 32 |
共晶结构还显示化合物32的喹唑啉酮环与异丙基苯环的构象呈垂直取向, 由于邻位的双取代造成的阻转异构, 存在光学异构体, 经色谱拆分和测定活性发现R-32为优映体, 抑制细胞活性的IC50 = 0.130 μmol·L-1, 显著高于S-异构体。
32的共晶结构还提示了新的信息: ①在哌嗪的C2位置有可与Cys12、Glu62和Tyr96接触的空间; ② R-构型末端的氟代苯酚处, 还有空间可加入与Val9和Ile100结合的疏水性基团, 以及与Arg68和Asp69的极性基团的结合空间。这些都是进一步优化32的契机。虽然32的水溶性很低, 而且过膜性和动物生物利用度也差, 但却是通往成药的最后的里程碑分子。
3.2 结构优化对R-32作结构优化, 目标是提高活性的同时, 对G12C变异细胞株的选择性抑制, 因而用发生G12S变异的A539细胞作为评价脱靶的量度, 同时还评价化合物的物化性质和药代行为, 结构变化的位置是喹唑啉酮的7位引入较大的疏水基团, 以增强与该空间的疏水性结合。同时在哌嗪环的2'位作甲基取代探索对活性的影响。此外还考察母核的8位元素CH作N等排变换的作用, 合成的化合物的结构与活性列于表 4, 物化性质和药代参数见表 5。
| Table 4 Optimization of the lipophilic "Tail" (R1) and piperazine substituent (R2) of (R)-32. a: Concentration for inhibition of 50% MIA PaCa cell growth through inhibition of extracellular-regulated kinase 1/2 after 2 h incubation. b: Viability assessed at 72 h by CellTiter-Glo luminescence assay (Promega) in MIA PaCa cells mutant at G12C. c: Viability assessed at 72 h by CellTiter-Glo luminescence assay (Promega) in A549 cells mutant at G12S |

| Table 5 Physico-chemical and pharmacokinetic properties of the modified compounds. a: Hepatic cells, μL·min-1 (106 cells). b: MDCK cell permeability A—B, μcm·s-1. c: Solubility in phosphate buffer saline in μmol·L-1. d: Solubility in fasted state simulated intestinal fluid in μmol·L-1 |
表 4和表 5的构效关系综合讨论如下: ①末端苯基(32) 换作萘基(33) 或吲唑基(34) 虽然提高了抑制细胞的活性, 但吸收性和生物利用度都较低, 所以不宜更换成大的疏水片段。②为提高过膜性将32的酚羟基去除, 35仍然保持细胞活性, 而且提高了过膜性(MDCK) 和溶解性。尤其是在哌嗪环上引入2'-甲基的36还提高了小鼠的口服生物利用度。③ 36的氟原子用亲脂性强的氯(37) 或三氟甲基(38) 取代, 活性下降, 也降低了过膜性, 所以是不利的。④ 40和41分别是32和39的喹唑啉酮环的8-氮杂电子等排体, 旨在使酚羟基与8-aza形成分子内氢键, 以降低羟基穿越细胞膜的去溶剂化的能量损耗, 结果表明, 40和41的过膜性显著提高, 而且溶解度也提高了。化合物41的生物利用度达到33%, 而且细胞活性因之提高(p-ERK IC50 = 44 nmol·L-1; MIA PaCa-2活力IC50 = 5 nmol·L-1)。⑤将41的取代基变换为42、43或44的氟、氯、溴, 活性和药代性质的综合效应都不如41, 提示结构的优化需要在活性和成药性之间作调整。
3.3 解决阻转异构的困惑化合物41的母核氮杂喹唑啉酮与异丙基苯的连键为手性轴, 是由于邻位的取代基形成阻转异构的缘故, 图 9是对映体转化的能量参数和光活体的活性。然而这一对异构体的构型不能完全稳固存在, 而是慢慢地互相转化, 这对确定药物疗效是个不利因素, 因为对映体的活性不同。例如41的光活体在25 ℃逐渐消旋化, 半衰期为8天, 转化自由能垒ΔG≠ = 26 kcal·mol-1, 所以这对于将R-41类的化合物研发成药物是个障碍。

|
Figure 9 Compound 41 atropisomers are not configurationally stable at 25 ℃ |
解决的办法是变换结构, 途径有3个: 一是提高构型转化的能垒, 使活化自由能ΔG≠提高到30 kcal·mol-1, 这样在室温下能以稳定的构型存在; 二是将转化能垒降低到20 kcal·mol-1, 成为容易转化的混合物, 研发成消旋体; 三是制备对称的取代基, 消除手性轴。
为此, 用两种方法研究具有阻转异构的化合物转换构型的能垒。对于慢速转化的化合物用1H NMR信号随时间的变化测定阻转异构体的比例的信息, 拟合艾林方程(Eyring equation) 求出活化自由能值ΔG≠; 快速转化的化合物用变温核磁谱(VT-NMR) 加以确定(Rotzler J, Gsellinger H, Bihlmeier A, et al. Atropisomerization of di-para-substituted propyl-bridged biphenyl cyclophanes. Org Biomol Chem, 2013, 11: 110-118)。合成的化合物结构、活性和转化活化能列于表 6。构效关系如下: ①将异丙基换成环丙基(45) 旋转能垒变化不大, 活性还略有所失; 换成叔丁基(46) 的构型稳定性显著增加, 但活性下降。②苯环上双取代的化合物47的细胞活性提高, 也提高了构型的稳定性(ΔG≠ > 30 kcal·mol-1); 将41的苯环换成异丙基吡啶的49, 基本维持了高活性, 但旋转能垒不高, 构型呈亚稳态(t1/2~5 h), 因而不适宜研发。③换成五元芳杂环的化合物(50~52) 由于不能固定构型, 也阻碍自由旋转, 故不可取。④苯环的2, 6位被二乙基(54) 或环丙基(55) 或异丙基(56) 取代的对称性化合物活性都很强, 而且也阻止了手性键的旋转。
| Table 6 Atropisomer stability and KRASG12C activity as a function of cryptic pocket arene identity and substitution pattern |



对5个高活性化合物评价药动学性质, 表 7的数据显示, 它们的半衰期、清除率和血浆蛋白结合率(或表征为游离的化合物的份额) 相差不多, 但对小鼠的口服生物利用度(F) 不同, R-41与51的F值相近, 而其他3个的吸收性差。化合物R-41小鼠灌胃10 mg·kg-1的最大血药浓度(Cmax) 是IC50的4.5倍, 51为1.5倍, 提示R-41的体内活性强于51。所以优选出R-41是高活性化合物中药代性质优良的化合物。
| Table 7 Mouse PK profilesa of compounds with atropisomer configurational properties suitable for development. a: iv/po dosing in BALB/c mice (iv, 1 mg·kg-1, DMSO; po, 10 mg·kg-1). b: Plasma protein binding by ultracentrifugation. c: Maximum unbound plasma concentration (Cmax·fu) |
化合物R-41的胜出是将苯并环换作吡啶并环, 提高了该片段与“神秘”腔穴的结合力, 为了提高其物化和生物药剂学性质, 对吡啶并环的氯与氟原子的变换, 苯环用吡啶环的变换, 合成的化合物结构与性质列于表 8。结果提示: ①将(R)-41的苯环的一个CH换成N原子, 57和58的水溶性显著提高, 对KRAS的活性没有改变, 但过膜性降低了, 测不出口服吸收度。②换成嘧啶环59也使溶解度提高、活性基本不变, 但过膜性和口服利用度显著降低。③只将(R)-41的氯原子改换成氟, 60溶解度提高两倍, 细胞活性略有下降, 但过膜性和口服吸收都显著降低。④化合物61~63的溶解性都提高, 61的细胞活性略有降低, 而过膜性和生物利用度都显著的提高, 是个突出的优化分子(Lanman BA, Allen JR, Allen JG, et al. Discovery of a covalent inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors. J Med Chem, 2020, 63: 52-65)。
| Table 8 Biopharmaceutical optimization of (R)-41 significantly enhances bioavailability |
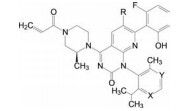
R-61的体内药效学表明, 灌胃移植MIA PaCa-2细胞的裸鼠模型, 在10 mg·kg-1剂量下显著抑制ERK的磷酸化, 进而评价了剂量-时间的药效/药代的关系, 以及对动物安全性试验, 证明R-61是选择性KRAS抑制剂, 经系统的临床前研究后, 定名为索托拉西(sotorasib, AMG510) 于2018年开始临床研究, 历时三年后基于II期开展的名为CodeBreaK100的临床研究, 对126例KRASG12C变异的中晚期肺癌在免疫疗法和化疗无效的患者应答率为37%, 中位应答时间10个月, 88%患者症状明显改善, 于2021年5月28日经美国FDA批准上市。
5 与KRAS的结合模式索托拉西与KRASG12C的共晶结构(图 10) 显示, 母核氮杂喹唑啉环占据了S-II疏水腔中, 丙烯酰胺与Cys12的巯基发生共价键结合。S-甲基哌嗪采取扭船式构象, 使2'-甲基与Cys12和Tyr96接触, 吡啶环上的异丙基完全进入S-II疏水腔中, 与Tyr96、His95和Gln99发生范德华作用。吡啶的N原子对手性抽的稳定性没有影响, R构型在25 ℃半衰期计算为t1/2 > 180年(ΔG≠转动能垒 > 31 kcal·mol-1)。

|
Figure 10 X-ray crystal structure of sotorasib bound to GDP-KRASG12C complex |
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
索托拉西于2021年5月28日美国FDA根据II期临床结果的安全有效性批准上市, 成为治疗携带有KRASG12C变异的中晚期非小细胞肺癌患者的首创性药物, 引起了全球业界的瞩目。KRAS是个致癌基因, KRAS蛋白被称作肿瘤跳动的心脏。KRAS容易发生变异 (G12C), 更加增强了增殖活力, 功能上使肿瘤成为永动机。而且, 大约30%的人类肿瘤有KRASG12C的变异, 显然成为研制的重要靶标。大约40年前科学家就已揭示KRAS变异与肿瘤的关系, 认定是重要的药物靶标, 但多年来的研究进展缓慢, 原因之一是该蛋白与底物GDP/GTP的结合力非常强, 达到皮摩尔水平, 因而很难找到于该结合腔同GDP竞争性结合的小分子抑制剂, 此外, KRAS没有明显的疏水腔或结合位点作为药物设计的抓手和切入点。安进 (Amgen) 公司迎难而上, 应用基于片段的药物设计 (FBDD) 方法和共价结合 (covalent binding) 原理, 找到了动态发生的变构结合位点, 将药物共价结合于Cys12上, 锁定KRAS为不可逆的非活化构象状态, 突破了KRAS非可药性靶标的思想束缚, 成为全球首创的以KRASG12C为靶标的小分子靶向药物, 在一定程度上具有划时代的意义。本文基于公开发表的文献, 简述了研制索托拉西的药物化学过程。 (编者按)