 2021, Vol. 56
2021, Vol. 56



“生物电子等排”(bioisosterism) 是由早期的“电子等排”(isosterism) 发展和延伸来的[1]。早在1909年James Moir就提出相对简单的化学实体之间的电子等排性概念, 这一概念又被H.G.Grimm的氢化物置换定律进一步完善, 10年后Irving Langmuir根据实验有效地证实上述电子等排性概念。即凡是具有相同数目的原子和电子, 并且电子排列状况也相同的分子、原子或基团(离子) 称为电子等排体。20世纪30年代Hans Erlenmeyer发表了一系列研究, 该研究扩展了Karl Landsteiner的早期工作。Erlenmeyer重氮离子与蛋白质反应衍生的人工抗原的研究表明, 抗体无法区分苯基和噻吩环或O、NH和CH2。在前辈的研究基础上, Harris Friedman于1950年提出了“生物电子等排”及“生物电子等排体”等新概念, 将生物电子等排体定义为引起类似生物效应的化合物, 同时指出化合物可能是电子等排体, 但不一定是生物电子等排体。这一概念预期生物电子等排体的应用将取决于生物效应, 而不依赖于物理化学性质。生物电子等排体通常不是精确的结构类似物, 而是在生物学特性上更相似的化合物。因此, 在一种生物化学环境中有效的生物电子等排体不一定能够适用于另一种生物化学环境, 往往需要针对特定情况仔细选择和裁剪生物电子等排体。
生物电子等排体的设计经常引起结构变化, 这些结构变化可能是有益的, 也可能是有害的, 因其大小、形状、电子分布、极化率、偶极矩、极性、亲脂性和pKa可能在分子中起着关键的作用。在当代药物化学研究中, 引入生物电子等排体的化合物的开发和应用是一种基本的药物设计策略, 有助于解决与候选药物的设计和开发相关的许多问题。借此可以提高分子的活性和选择性、改变物理性质、减少或改变新陈代谢、消除或修饰毒性代谢物以及获得新的知识产权。
在众多的含氟药物上市(图 1) 和有机氟化学蓬勃发展的今天, 对新的含氟基团的药学和化学探索越来越受到科学家们的重视, 本文扼要地综述二氟甲基(CF2H) 的理化性质、化学制备方法以及作为生物电子等排体在药物设计中的应用, 以便于给药物研发人员提供参考。

|
Figure 1 Examples of therapeutic drugs, herbicides, fungicides and agricultural chemicals with difluoromethyl group |
众所周知, 氟是电负性最强的非金属元素。将氟引入有机分子中产生其独特的化学和物理性质, 氟原子的高电负性使C-F键与蛋白质主链或侧链酰胺基形成偶极和多极相互作用, 或与精氨酸胍基的π面形成偶极和多极相互作用。虽然氟可以提供独特的蛋白质-配体相互作用, 但将小分子配体优化为候选药物需要平衡药效与物理化学和药动学性质。
由于C-F键强度强于C-H键, 用氟代替氢已成为多年来降低细胞色素P450 (CYP) 介导代谢的药物化学策略之一, 并取得了许多成功的例子。氟可以通过阻断修饰位点直接减少代谢, 也可以通过改变代谢位点附近的电子云密度间接地减少氧化代谢。根据添加氟对整体分子性质的影响, 如亲脂性增加和氟化位点与代谢位点的电子和空间关系, CYP介导的氧化速率可以降低或提高。与氢相比, 氟原子的大小和电负性都有所增加, 这对分子构象有实质性的影响, 通过配体重组改变其结合构象所需的能量来影响与靶点的结合亲和力。含有OCH3和OCF3的分子在药物化学项目中已得到了很好的探索, 而对含有OCHF2和OCH2F基团的分子的研究逐步受到更多的重视。含有OCF2H基团的分子表现出可变的亲脂性(πx = + 0.2~0.6), 通过围绕O-CF2H键的简单键旋转(g = 0.45 kcal·mol-1), 它们可以根据周围的化学环境不同而改变其亲脂性(图 2)。与CF3和OCF3相比, OCF2H基团具有较低的亲脂性, OCF2H基团可以作为具有不同亲脂性的氢键供体或接受体, 从而丰富分子与结合口袋中残基的相互作用。因此, 将CF2H基团引入到药物和农药中, 可以微调它们在体内的吸附、分布、代谢、排泄和毒性(ADMET) 特性[2, 3]。

|
Figure 2 Physicochemical properties of fluoromethyl compounds |
二氟甲基上的氢是一个相对较弱的氢键(HB) 供体。然而, 各种不同功能团(FG) 对其形成氢键能力有影响, 化合物的A值(HB酸度) 在0.035到0.165之间。同时二氟甲基上的氟又可以是一个氢键(HB) 的接受体(图 3、4)。二氟甲基与芳基或烷基相联, 其亲脂性低于相应的CH3和CF3取代化合物。二氟甲基与亚砜或砜α位置相联表现出最高的A值, 但会导致亲脂性显著增加, 可能是由于化合物的极性降低以及所附的SO或SO2基团的HB接受体能力降低, 同时增加溶质体积。在烷基醚中, 以α位置的CH3被CF2H取代后, 导致亲脂性显著增加, 这可能是由于这些化合物的HB接受氢能力降低所致, 但CF2H比CF3亲脂性低。XCF2H的氢键酸度与苯胺/胺或硫酚相似, 而低于苯酚/醇。与苯甲醚或苯硫甲醚母体化合物相比, 两个系列的亲脂性可能改变, 是否增加或减少取决于芳香环上取代基的类型。

|
Figure 3 Physicochemical properties of O, S-difluoromethyl compounds |

|
Figure 4 The effect of functional groups (FG) on the ability of CF2H to form hydrogen bonds |
二氟甲基与芳基或烷基相联, 其亲脂性低于相应的CH3取代化合物, 因此, CF2H可视为亲脂性较低的CH3生物电子等排, CF2H也比CF3亲脂性更低。二氟甲基与芳基或烷基相联, 其亲脂性、体积大小以及氢键酸度接近于的SH取代化合物, 因此, CF2H可视为亲脂性相似的SH生物电子等排体。二氟甲基与芳基或烷基相联, 其亲脂性强于OH取代化合物, 因此, CF2H可视为亲脂性较高的OH生物电子等排体。二氟甲基与芳基或烷基相联, 其亲脂性强于NH2取代化合物, 因此, CF2H可视为亲脂性较高的NH2生物电子等排体。二氟甲基硫基(-SCF2H) 是亲脂性氟烷基三氟甲基硫基(-SCF3) 的生物电子等排体。然而, -SCF2H的性质与-SCF3有很大不同。-SCF3的Hansch参数pR = 1.44, 而二氟甲硫基的Hansch参数pR = 0.68。-SCF2H的亲脂性仅略高于甲基(pR = 0.56), 远低于三氟甲硫基。这种亲脂性变化以及结构上的细微变化为药物化学家提供了一个微调药物分子亲脂性的机会。另一方面, 含二氟甲基的二氟甲硫基, 通常被认为是OH或NH2基的生物电子等排体, 可以提高药物分子的结合选择性(表 1)。
| Table 1 Bioisosteres related to difluoromethyl group |
上述发现将有助于设计氟化药物候选物, 提高代谢稳定性或结合亲和力, 以及合理调节重要药物特性的能力。CF2H基团性质不仅受到与其相联的基团影响, 同时CF2H基团也会对其相联的基团产生很大的影响[4-6]。
考虑到上述结果以及Linclau等[4]之前的研究, 可以得出这样的结论: CF2H基团除了作为一个功能团依赖性HB供体, 也可以调节分子的亲脂性(logPoct)。通过影响主要参数如极性、HB碱度和分子体积等。因此, 进一步研究CF2H基团在不同分子环境下对上述性能的影响仍然是有必要的。
3 二氟甲基在药物设计中运用 3.1 提高生物活性吡唑类杀菌剂2的CHF2部分与邻近的C=O之间的分子内氢键相互作用已被红外光谱和核磁共振氢谱数据证实(图 5)。该类化合物能抑制真菌琥珀酸脱氢酶, 这种相互作用似乎对2的杀菌活性很重要, 因为缺少氢键供体的CF3同系物1显示出较弱的生物学效应[7]。3-(二氟甲基)-1-甲基-1H-吡唑-4-甲酰胺部分是一系列杀菌剂中常见的结构骨架, 包括氟吡唑、赛的松和异吡唑(图 1)。

|
Figure 5 The replacement of CF3 with CF2H to form an intramolecular hydrogen bond, which increases the activity by 4 times |
最近, 在单晶X射线结构研究中, 发现CHF2部分和相邻分子的腈之间形成分子间的氢键。噁二唑衍生物3的键角(140o) 和键长(2.48 A) 如图 6所示[8]。

|
Figure 6 Intermolecular H-bond observed in the single crystal X-ray structure of oxadiazole 3 |
二氟甲基作为醇和硫醇的生物电子等排体在药物设计中得到广泛应用, 其中CHF2部分被用作氢键供体, 而用C-F替代C-OH(SH) 则依赖于两者相近的极性。
CHF2部分可作为氢键供体, 与HCV NS3蛋白酶结合是最引人注目的成功应用, 在设计一种HCV NS3蛋白酶抑制剂(基于六聚肽的NS3抑制剂) 中, 应用二氟甲基代替天然底物p1位点的半胱氨酸SH残基, 其结构相似性如图 7中的简单方式所示[9]。

|
Figure 7 The similarity diagram of RSH (4) and RCF2H (5) bound to p1 residues |
HCV NS3/4a类抑制剂的设计灵感来源于先前吡唑2的研究结果, 发现8与Cys模拟物6具有同等生物活性, 表明具有良好的生物电子等排体功能。其甲基类似物7的活性降低了20倍(图 8), 进一步证实了8结构中存在H-键供体的重要性[10]。

|
Figure 8 Design of HCV NS3/4a inhibitors |
Zheng等[11]设计和合成了一种含有二氟甲基环丙基氨基酸的丙型肝炎病毒NS3蛋白酶三肽酰磺酰胺抑制剂10 (图 9)。10与NS3/4a蛋白酶复合物的共晶结构显示CHF2部分的C-H与酶的LEU135的主链羰基之间形成H键。结构-活性关系(SAR) 研究表明, 这种H-键将酶抑制效力提高13倍以上。

|
Figure 9 10a: The co-crystal structure of HCV NS3/4a and 10; 10b: The H-bonding site of NS3/4a protease complex p1 subside and 10 |
在HCV NS3蛋白酶抑制剂设计中, 在环丙基上引入CHF2而获得高效抑制剂(图 10), 如glecaprevir (11) 和voxilaprevir (12), 这两个药物都表现抗泛基因型病毒活性, 并且已被FDA批准上市[12]。

|
Figure 10 The FDA-approved anti-hepatitis C drugs containing CF2H group |
溶血磷脂酸(LPA) 13对GPCR四种受体有结合作用(定为LPA1-4), 也作为核激素受体-过氧化物酶体增殖物激活受体γ (PPARγ) 的激动剂。然而, 由于脂多糖易于通过酰基迁移进行重排到末端, 故将二氟甲基取代末端醇羟基(OH), 设计并合成二氟化脂多糖类似物14 (图 11), 虽然14未能与脂多糖LPA1-3受体相互作用, 但这种化合物保留PPARγ受体激动剂的活性。这一实例也表明生物电子等排体实际应用时的局限性[13, 14]。

|
Figure 11 Lipopolysaccharide 13 and lipopolysaccharide difluomethyl analogue 14 |
CHF2和OH基团之间的生物等效性已扩展到羟肟酸类化合物(图 12), 其中, 在5-脂氧合酶抑制剂15氮上的OH被CHF2基团取代后获得16, 能保留其5-脂氧合酶抑制活性[15-17]。

|
Figure 12 The design of 5-lipoxygenase inhibitors |
在某些情形下, 用二氟甲氧基取代甲氧基不仅可以提高代谢稳定性, 而且还能提高生物活性。Zhang等[18]在研究NS4B抑制剂的构效关系时发现, 在吲哚环的6位引入体积较小的亲脂性基团OCHF2 (18), 比OCH3 (17) 的抗丙肝病毒的活性明显提高10倍(图 13)。

|
Figure 13 NS4B inhibitors 17, 18 |
Yokokawa等[19]在研究抗结核病药物中发现虽然化合物19和20对结核分枝杆菌(MTB) 抑制活性没有改变(图 14), logP的测量值也略有增加, 但CF2H取代化合物20的水溶性比CF3同系物19高20倍以上。

|
Figure 14 MTB inhibitors 17, 18 |
临床前研究发现21是一种有效的组织蛋白酶K抑制剂(IC50 = 0.3 nmol·L-1), 其对组织蛋白酶B (IC50 = 1 034 nmol·L-1)、L (IC50 = 2 295 nmol·L-1) 和S (IC50 = 60 nmol·L-1) 具有良好的选择性(图 15), 但是该候选物的高结晶度导致水溶性较低, 口服生物利用度较差。为了解决溶解度差引起生物利用度问题, 设计并合成了CHF2取代的类似物, 增加了碱度(pKa增加~1单位), 使易与磺酸和HCl形成盐, 并导致分子的亲脂性降低。化合物22不仅保留了与21相似的有效的组织蛋白酶抑制活性(IC50 = 0.8 nmol·L-1) 和蛋白酶选择性, 而且对大鼠口服生物利用度得到明显的提高[20]。

|
Figure 15 Cathepsin K inhibitors 21, 22 |
Tang等[21]在研究病毒衣壳抑制剂中发现, 用代谢更稳定的CF3生物电子等排体取代甲基导致抗病毒活性下降了6倍(图 16), 这可能是由于完全丧失C-H键, 导致失去其σ*轨道和蛋白质中的π体系的超共轭效应(hyperconjugation)。这种超共轭是甲基效应的分子基础。为了恢复其超共轭效应, 将CF3改为CHF2生物电子等排体, 由此产生的类似物25不仅显著提高了口服生物利用度(F = 25%), 也增强了抗病毒活性(图 16)。

|
Figure 16 The leader optimization of the viral capsid inhibitors |
有关研究表明, 在化合物结构优化过程中, 综合考量亲脂性和分子量, 并在不影响膜渗透性的情况下, 提高代谢稳定性, OCHF2比OCH3和OCF3更具有优势[22]。
表 2中汇总了促肾上腺皮质激素释放因子-1 (CRF-1) 抑制剂26~29系列的活性和清除率。数据表明含有OCHF2基团的化合物抑制活性更强和清除率更低[23]。
| Table 2 Activity and clearance rate of CRF-1 inhibitors 26-29 series |
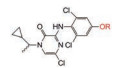
辉瑞公司内部数据库显示, 在优化ADME性能方面, PhOCF2H可能比PhOCF3有明显优势。表 3数据表明, 与甲氧基和三氟甲氧基相比, 二氟甲氧基化合物32和34具有更低的HLM清除率和更高的犬肾细胞膜渗透性[1]。
| Table 3 HLM clearance and canine renal cell membrane permeability. a: Human liver microsome clearance; b: Ralph Roth canine kidney cell membrane permeability |
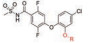
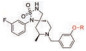

通过阻断非芳香族代谢氧化来防止药物的生物活化是目前常用的一种结构优化手段。在PDE-IV抑制剂研究领域, 双酚醚是一种常见的结构特征(图 17)[24, 25]。先导化合物(36) 可被代谢氧化脱烷基再氧化成高反应性的醌类形式(38/39), 后者可与内源化合物发生共价结合从而产生毒性。药物化学方面想努力消除这种潜在的生物活化。用二氟甲基醚取代烷基醚得到化合物(37), 导致体外代谢的大幅度降低, 并阻止共价结合。值得注意的是, 二氟甲氧基随后出现在许多文献中, 包括roflumilast (40), 一种2011年被FDA批准用于治疗COPD恶化的化合物[26]。

|
Figure 17 The replacement of alkyl ether (36) with difluoromethyl ether to block the metabolic site |
Peleg等[27]使用生物电子等排体方法, 开发了具有较高抗增殖活性且较低钙血症的天然激素1α, 25-二羟基维生素D3 (41) 的二氟甲基化类似物43 (图 18)。天然激素会引起高钙血症毒性, 因此, 研究者们一直在努力研究, 达到最大程度发挥1α, 25-二羟基维生素D3治疗潜力的同时, 最大程度减少高钙血症毒性。将维生素D3上1α-OH改成CH2OH, 再进一步改变成新的二氟甲基化(CF2H) 类似物, 结果将毒性降低了20倍。不可否认, 二氟甲基化类似物对人类维生素D受体的亲和力下降了一些, 这可能是由于二氟甲基上氢与受体形成氢键较弱所致。然而, 通过引入增强的侧链克服了这一问题。因此, 发现新的类似物43与天然激素具有相似的转录活性, 在体外具有很强的抗增殖作用, 而体内的钙化活性却很小。

|
Figure 18 The replacement of OH (41) with CF2H (43) to reduce the toxic hypercalcemia |
Zafrani等[28]报道了一种以一溴二氟甲基膦酸二乙酯(45) 为二氟卡宾前体的苯酚和硫代酚二氟甲基化的简便方法(图 19)。这种市售的膦酸盐在碱性水解(-78 ℃到室温) 时会发生P-C键断裂, 从而产生溴二氟甲基阴离子, 该阴离子随后转化为二氟卡宾中间体。后者与酚类化合物44a或硫代酚类化合物44b反应, 得到相应的二氟甲基醚和硫醚, 产率较高。由此产生的环保的副产物磷酸二乙酯离子, 由于其在水中具有良好的溶解性, 很容易从反应混合物中分离出来。由于反应条件温和, 可用于羰基或烯醇功能基的酚酸盐离子选择性的二氟甲基化。

|
Figure 19 Preparation of difluoromethyl anisole and thioanisoles |
Zhang等[29]在2009年报道了N-对甲苯基-S-二氟甲基-S-苯磺胺与几种亲核试剂反应, 特别是硫代亲核试剂(图 20)。在60 ℃温度条件下, 可与不同基团取代的各种硫代酚进行反应, 包括脂肪族和芳杂硫醇都能较好的进行(48g和48h)。尽管该试剂可能被视为“CF2H+”等效物, 但作者证明其作为二氟卡宾中间体发挥作用。

|
Figure 20 Preparation of difluoromethyl thioaromatic ethers and alkane ethers (Ⅰ) |
同一研究组后来又报道了二氟甲基(正丁基) 氯化铵与硫醇衍生物的反应, 该反应是在NaH存在下, 在温和的反应条件下(室温), 获得二氟甲基硫代芳香醚50 (图 21), 产率较好[30]。Bayarmagnai等[31]提出了一种将SCF2H直接引入分子的策略, 为了解决现有二氟甲硫基化试剂的缺乏问题, 采用TMSCF2H进行了铜介导的二氟甲基硫醚化一锅煮反应, 反应试剂CuCF2H不经分离直接用于取代反应。其中TMSCF2H可从TMSCF3来制备。

|
Figure 21 Preparation of difluoromethyl thioaromatic ethers and alkane ethers (Ⅱ) |
Zhu等[32, 33]以价廉的商品化邻苯二甲酰亚胺为原料, 经四步反应合成了一种亲电性二氟甲基硫酰亚胺试剂(51)。在Lewis酸Me3SiCl或者CuI的存在下, N-二氟甲基硫代邻苯二甲酰亚胺能够有效地将二氟甲基硫代基引入到多种亲核试剂中, 包括胺、硫醇、β-酮酸酯、芳基/乙烯基硼酸、炔烃和富电杂环芳烃, 如氧化吲哚、吡咯-1H吡咯[2, 3-b]吡啶、咪唑[1, 2-a]吡丁胺、氨基噻唑、异恶唑(图 22~25)。这些反应底物多样性和官能团良好耐受性使N-二氟甲基硫代邻苯二甲酰亚胺在制备更复杂、功能团更密集的药物分子方面显示巨大潜力。

|
Figure 22 Reactions of difluoromethyl sulfimide reagent |

|
Figure 23 The direct cross-coupling reaction with electric-rich heterocyclic aromatic compounds (Ⅰ) |

|
Figure 24 The cross-coupling reaction with boronic acid substituted aromatic compounds (Ⅱ) |

|
Figure 25 The cross-coupling reaction with aliphatic active sites |
有关制备二氟甲基醚/硫醚的方法较多, 这里就不逐一叙述了, 可参见Xiong等[34, 35]文献综述。
4.2 二氟甲基与烷基相连接的化合物合成硫原子在形成CF3基团的化学过程中也起着重要的作用。因此, 在碱性条件下, 2-烷基-1, 3-二硫烷衍生物66 (可以从醛制备) 很容易被溴化物烷基化, 随后可以与BrF3反应, 形成相应的1, 1-二氟甲基烷烃(68, 71), 产率为60%~75% (图 26)。该反应适用于一级和二级卤代烷, 二硫烷的两个硫原子对反应至关重要[36]。

|
Figure 26 Reaction of BrF3 with 1, 3-disulfane derivatives |
自2015年首次制备CF2HCHN2 (72) 以来, 在有机合成方法中有多种不同的应用, 目前已经报道了三种不同类型的反应: a) 偶极[3+2]-环加成反应; b) 卡宾反应; c) 酯化反应(c-亲电)。
CF2HCHN2 (72) 不稳定、易于分解, 因此该试剂常常以二氟乙胺为原料, 经氧化后在反应液产生, 不经分离直接参与反应。Mykhailiuk[37]报告了该试剂与烯烃的偶极[3+2]环加成反应(图 27), 该反应提供了直接获得CHF2取代的吡唑啉的合成方法。研究表明, 缺电子的烯烃, 如马来酰亚胺、丙烯酸酯或烯酮可以顺利地与CF2HCHN2 (72) 发生环加成反应。而富电子的烯烃, 如苯乙烯和烯醇醚却不能反应。

|
Figure 27 Dipole [3+2]-cycloaddition reaction of CF2HCHN2 with olefins |
如前所述, 二氟二氮乙烷72是一种易发生分解反应的敏感试剂。因此, Duan等[38]开发了一个比较稳定的试剂76, 可以取代二氟二氮乙烷(图 28)。在锌还原剂和FeTPPCl催化剂的存在下, 二氟甲基磺酸盐(76) 可以很容易地产生二氟甲基卡宾, 再与苯乙烯反应, 可高选择性地产生反式二氟甲基取代的环丙烷化合物。

|
Figure 28 [1+2]-cycloaddition reaction of (2, 2-difluoroethyl)diphenyl-l3-sulfane with alkenes |
Hayashi等[39]于2002年首次报道2, 2-二氟-1, 3-二甲基咪唑啉(82, DFI) 为一种新型的氟代试剂(图 29)。在85 ℃下, 直接与不可烯醇化的醛发生反应, 产生二氟甲基取代的芳环化合物71, 收率较高。目前已开发出多个将芳香醛基转变成二氟甲基的三氟硫代试剂(图 29, 82~85)[39-44], 反应通常在低温或者室温进行。二氟甲基还可以通过C-C偶联反应直接引入到芳香环或芳香杂环上(图 29, 反应条件5~7)[43, 44]。其中Xu等[45]报道以(dppf)Ni(COD) 作为催化剂, 在室温条件下, 锌试剂(DMPU)2Zn(CF2H)2与卤代芳烃芳进行C-C偶联反应。

|
Figure 29 Preparation method of difluoromethyl aromatics (Ⅰ) |
最近, Tung等[46]开发出一种方便的二氟甲基取代的芳环化合物的方法, 使用现存的二氟乙酸作为二氟甲基化试剂, 直接进行杂芳族化合物的C-H二氟甲基化(图 30), 通过调整反应温度可控制单-二氟甲基化与双-二氟甲基化产物, 缺点是只有芳杂环氮邻位比较活泼的C-H键易发生C-C偶联反应。

|
Figure 30 Preparation method of difluoromethyl hetro-aromatics (Ⅱ) |
Mykhailiuk[47]首次报道CF2HCHN2与炔烃发生[3+2]偶极环加成(图 31), 可获得产率较好的二氟甲基吡唑类化合物(85a~85c)。这种一锅法很容易放大到克级的吡唑制备。

|
Figure 31 Dipole [3+2]-cycloaddition reaction of CF2HCHN2 with alkynes |
Khutorianskyi等[48]首次以二氟乙酸乙酯为原料, 在反应体系内, 经两步合成了一种新型化学试剂—二氟甲基一氧化氮CF2HCNO (87, 图 32)。该试剂不经分离直接与炔烃、烯胺发生[3+2]-环加成反应, 生成含CF2H基团取代的异恶唑类化合物88。

|
Figure 32 Dipole [3+2]-cycloaddition reaction of CF2HCNO with alkynes |
Li等[49]通过使用二氟甲烷亚磺酸锌[DFMS, (CF2HSO2)2Zn, Baran试剂], 通过类似的自由基过程, 实现了铁催化的芳基取代的丙烯酸的二氟甲基化(图 33)。该方法仅适合于富电子芳基取代丙烯酸的二氟甲基化并获得以反式为主的产物90a~90d。

|
Figure 33 Iron-catalyzed decarboxylative difluoromethylation of cinnamic acids with zinc difluoromethanesulfinate |
东京理工大学创新研究所化学与生命科学实验室的Arai等[50]开发出一种新颖而简单的方法, 通过可见光直接将CF2H基团引入到有机骨架中(图 34、35)。该反应属于光氧化还原催化。在可见光照射(425 nm蓝光LED灯) 及光催化剂fac-[Ir(ppy)3] (ppy = 2-pyridylphenyl) 存在下, 以N-对甲苯基-S-二氟甲基-S-苯硫氧基胺(91) 为CF2H基团源, 与含不同官能团的烯烃及水、醇、羧酸等亲核剂进行反应, 可选择性得到β-CF2H取代的醇、醚和酯类化合物93、94。

|
Figure 34 Photo-redox catalytic reaction of N-p-tolyl-S-difluoromethyl-S-phenylsulfoxyamine |

|
Figure 35 Examples of photo-redox catalytic reaction of N-p-tolyl-S-difluoromethyl-S-phenylthiolamine with alkenes |
Shen等[51]开发一种制备CF2H取代的醇类化合物的方法, 以环氧化合物(3, 4, 5元环) 为起始原料, 化合物95为CF2H源, 经多步反应而制得CF2H取代的醇(图 36、37)。

|
Figure 36 Examples of the reaction of 95 with epoxy compound |

|
Figure 37 The preparation of 100 using 95 as difluoromethyl source |
首先用TBSCl和咪唑在DMF中处理化合物(98), 反应过夜, 得到90%产率的磺胺95, 在Lews酸BF3和LiHMDS存在下, 磺胺95与环氧化合物99低温下反应, 12 mol·L-1 HCl后处理后产生中间体97d。最后在金属镁和催化量二溴乙烷存在下, 室温反应制备CF2H取代的醇100 (图 37)。其中3, 4元环氧烷底物反应收率较高。
4.5 二氟甲基(CF2H) 硫酯类化合物的制备Guo等[52]报道一种制备二氟甲基(CF2H) 硫酯类化合物的新方法, 该法以廉价、易得的醛(芳香醛、脂肪族醛和α, ß-不饱和醛) 为原料, S-(二氟甲基) 苯磺酸酯为反应试剂(SCF2H源), 在氧化剂-叔丁基过氧化氢(TBHP) 存在下, 合成了二氟甲基硫酯102 (图 38)。该方法反应条件温和, 官能团耐受性好, 底物范围宽, 特别是不含金属, 有可能成为后期合成中间体的重要方法。

|
Figure 38 Examples of the reaction of S-(difluoromethyl)benzenesulfonate with aldehyde 101 |
目前已报道了多种方法可以获得N-二氟甲基含氮杂环化合物[53-56]。氯代二氟甲烷是一种氯氟碳气体, 是常用的试剂。但在近几年来, 已经开发出一些实用性较强的试剂(图 39, 反应条件2~5), 由此可以获得二氟甲基卡宾活性中间体, Huang等[54]使用TMSCF2Br和N-对甲苯基-S-二氟甲基-S-苯基磺氧基作为二氟甲基的来源, 借此在反应液中产生二氟甲基卡宾活性中间体, 不经分离直接与含氮杂环反应。Prakash等[55]也在中性条件下, 使用市售TMSCF3对咪唑类化合物进行二氟甲基化, 反应温度较高, 但也可以通过微波加热来缩短反应时间。

|
Figure 39 Examples of difluoromethylation reactions on the nitrogen of heterocyclic compounds |
Mao等[56]报告了一种市售的且稳定的二氟甲基化试剂BrCF2P(O)(OEt)2 (图 39, 反应条件2), 它可作为二氟卡宾前体, 能与含氮杂环化合物如咪唑和吡唑反应, 制备N-二氟甲基化的衍生物。该方案的显著特点包括反应条件温和、底物范围广和合成简单。
Polley等[57]报道了用市售、廉价、空气和水分稳定的溴二氟乙酸乙酯作为CF2H源, 在氢氧化锂促进下, 可以在甲苯磺酰基保护的胺类化合物上引入二氟甲基(图 40)。这方法的收率较好, 底物适用性较广。

|
Figure 40 Substrate scope for difluoromethylation of N-tosyl amines |
由于二氟甲基的特殊理化性质, 而且化学家已开发出制备含二氟甲基类的化合物的众多合成方法, 在未来合成方法学方面, 光催化引入二氟甲基领域需要进一步的研究, 未活化的芳烃的光催化二氟甲基化反应尚未开发, 具有一定挑战性。另一个值得特别关注的领域是应用流动系统以高收率和最短反应时间实现二氟甲基取代。
二氟甲基作为药物设计中的一种生物电子等排体策略, 在药物化学中得到越来越多的运用。伴随着含有二氟甲基的药物越来越多, 掌握何时以及如何在复杂的有机分子中引入二氟甲基, 深入地了解二氟甲基引入对分子生物活性和理化性质的影响, 有可能为新药发现提供更多的成功机会, 预计对二氟甲基的有机化学和药物化学的深入研究将继续受到重视[58, 59]。
作者贡献: 张寅生负责文献检索、分析、数据整理以及论文撰写、修改与检查。
利益冲突: 本文无利益冲突。
| [1] |
Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design[J]. J Med Chem, 2011, 54: 2529-2591. DOI:10.1021/jm1013693 |
| [2] |
Xing L, Blakemore DC, Narayanan A, et al. Fluorine in drug design: a case study with fluoroanisoles[J]. ChemMedChem, 2015, 10: 715-726. DOI:10.1002/cmdc.201402555 |
| [3] |
Zafrani Y, Yeffet D, Sod-Moriah G. The difluoromethyl bioisoster: examining the"lipophilic hydrogen bond donor" concept[J]. J Med Chem, 2017, 60: 2797-2804. |
| [4] |
Linclau B, Peron F, Bogdan E, et al. Intramolecular OH…fluorine hydrogen bonding in saturated, acyclic fluorohydrins: the g-fluoropropanol motif[J]. Chem Eur J, 2015, 21: 17808-17816. DOI:10.1002/chem.201503253 |
| [5] |
Sessler CD, Rahm M, Becker S. CF2H, a hydrogen bond donor[J]. J Am Chem Soc, 2017, 139: 9325-9332. DOI:10.1021/jacs.7b04457 |
| [6] |
Zafrani Y, Sod-Moriah G, Yeffet D. CF2H, a functional group dependent hydrogen bond donor: Is it a more or less lipophilic bioisostere of OH, SH and CH3?[J]. J Med Chem, 2019, 62: 5628-5637. DOI:10.1021/acs.jmedchem.9b00604 |
| [7] |
Erickson J, McLoughlin JI. Hydrogen bond donor properties of the difluoromethyl group[J]. J Org Chem, 1995, 60: 1626-1631. DOI:10.1021/jo00111a021 |
| [8] |
Goldberg K, Groombridge S, Hudson J, et al. Oxadiazole isomers: all bioisosteres are not created equal[J]. MedChemComm, 2012, 3: 600-604. DOI:10.1039/c2md20054f |
| [9] |
Narjes F, Koehler KF, Koch U, et al. A designed P1 cysteine mimetic for covalent and noncovalent inhibitors of HCV NS3 protease[J]. Bioorg Med Chem Lett, 2002, 12: 701-704. DOI:10.1016/S0960-894X(01)00842-3 |
| [10] |
Di Marco S, Rizzi M, Volpari C, et al. Inhibition of the hepatitis C virus NS3/4A protease: the crystalstructures of two protease-inhibitor complexes[J]. J Biol Chem, 2000, 275: 7152-7157. DOI:10.1074/jbc.275.10.7152 |
| [11] |
Zheng B, D'Andrea SV, Sun LQ. Potent inhibitors of hepatitis C virus NS3 protease: employment of a difluoromethyl group as a hydrogen-bond donor[J]. ACS Med Chem Lett, 2018, 92: 143-148. |
| [12] |
Martinez MD, Luna L, Tesio AY. Antioxidant properties in a non-polar environment of difluoromethyl bioisosteres of methyl hydroxycinnamates[J]. J Pharm Pharmacol, 2016, 68: 233-244. DOI:10.1111/jphp.12507 |
| [13] |
Xu Y, Prestwich GD. Concise synthesis of acyl migrationblocked 1, 1-difluorinated analogues of lysophosphatidic acid[J]. J Org Chem, 2002, 67: 7158-7161. DOI:10.1021/jo0203037 |
| [14] |
Xu Y, Qian L, Pontsler AV, et al. Synthesis of difluoromethyl substituted lysophosphatidic acid analogues[J]. Tetrahedron, 2004, 60: 43-49. DOI:10.1016/j.tet.2003.11.001 |
| [15] |
Chowdhury MA, Abdellatif KRA, Dong Y, et al. Synthesis of celecoxib analogues possessing an difluoromethyl-1, 2-dihydropyrid-2-one 5-lipoxygenase pharmacophore: biological evaluation as dual inhibitors of cyclooxygenases and 5-lipoxygenase with anti-inflammatory activity[J]. J Med Chem, 2009, 52: 1525-1529. DOI:10.1021/jm8015188 |
| [16] |
Yu G, Praveen Rao PN, Chowdhury MA, et al. Synthesis and biological evaluation of N-difluoromethyl-1, 2-dihydropyrid-2-one acetic acid regioisomers: dual inhibitors of cyclooxygenases and 5-lipoxygenase[J]. Bioorg Med Chem Lett, 2010, 20: 2168-2173. DOI:10.1016/j.bmcl.2010.02.040 |
| [17] |
Knaus EE. Synthesis of 1-(methanesulfonyl- and aminosulfonylphenyl)acetylenes that possess a 2-(N-difluoromethyl-1, 2-dihydropyridin-2-one) pharmacophore: evaluation as dual inhibitors of cyclooxygenases and 5-lipoxygenase with anti-inflammatory activity[J]. Bioorg Med Chem Lett, 2009, 19: 584-588. DOI:10.1016/j.bmcl.2008.12.066 |
| [18] |
Zhang N, Zhang X, Zhu J, et al. Structure-activity relationship (SAR) optimization of 6-(indol-2-yl)pyridine-3-sulfonamides: identification of potent, selective, and orally bioavailable small molecules targeting hepatitis C (HCV) NS4B[J]. J Med Chem, 2014, 57: 2121. DOI:10.1021/jm401621g |
| [19] |
Yokokawa F, Wang G, Chan WL. Discovery of tetrahydropyrazolopyrimidine carboxamide derivatives as potent and orally active antitubercular agents[J]. ACS Med Chem Lett, 2013, 4: 451-455. DOI:10.1021/ml400071a |
| [20] |
Isabel E, Mellon C, Boyd MJ. Difluoroethylamines as an amide isostere in inhibitors of cathepsin K[J]. Bioorg Med Chem Lett, 2011, 21: 920-923. DOI:10.1016/j.bmcl.2010.12.070 |
| [21] |
Tang J, Huber AD, Pineda DL, et al. 5-Aminothiophene-2, 4-dicarboxamide analogues as hepatitis B virus capsid assembly effectors[J]. Eur J Med Chem, 2019, 164: 179-192. DOI:10.1016/j.ejmech.2018.12.047 |
| [22] |
Xing L, Blakemore DC, Narayanan A, et al. Fluorine in drug design: a case study with fluoroanisoles[J]. ChemMedChem, 2015, 10: 715-726. DOI:10.1002/cmdc.201402555 |
| [23] |
Hartz RA, Ahuja VT, Rafalski M. In vitro intrinsic clearance-based optimization of N3-phenylpyrazinones as corticotropin-releasing factor-1 (CRF1) receptor antagonists[J]. J Med Chem, 2009, 52: 4161-4172. DOI:10.1021/jm900302q |
| [24] |
Chauret N, Guay D, Li C, et al. Improving metabolic stability of phosphodiesterase-4 inhibitors containing a substituted catechol: prevention of reactive intermediate formation and covalent binding[J]. Bioorg Med Chem Lett, 2002, 12: 2149-2152. DOI:10.1016/S0960-894X(02)00349-9 |
| [25] |
Guay D, Hamel P, Blouin M, et al. Discovery of L-791, 943: potent, selective, nonemeticandorally active phosphodiesterase-4 inhibitor[J]. Bioorg Med Chem Lett, 2002, 12: 1457-1461. DOI:10.1016/S0960-894X(02)00190-7 |
| [26] |
Tashkin DP. Roflumilast: the new orally active, selective phophodiesterase-4 inhibitor, for the treatment of COPD[J]. Expert Opin Pharmacother, 2014, 15: 85-96. DOI:10.1517/14656566.2013.837159 |
| [27] |
Peleg S, Petersen KS, Suh BC, et al. Low-calcemic, highly antiproliferative, 1-difluoromethyl hybrid analogs of the natural hormone 1R, 25-dihydroxyvitamin D3: design, synthesis, and preliminary biological evaluation[J]. J Med Chem, 2006, 49: 7513-7517. DOI:10.1021/jm0609925 |
| [28] |
Zafrani Y, Sod-Moriah G, Segall Y. Diethylbromodifluoromethyl-phosphonate: a highly efficient and environmentally benign difluorocarbene precursor[J]. Tetrahedron, 2009, 65: 5278-5283. DOI:10.1016/j.tet.2009.04.082 |
| [29] |
Zhang W, Wang F, Hu J. N-Tosyl-S-difluoromethyl-S-phenylsulfoximine: a new difluoromethylation reagent for S-, N-, and C-nucleophiles[J]. Org Lett, 2009, 11: 2109-2112. DOI:10.1021/ol900567c |
| [30] |
Wang F, Huang W, Hu J. Difluoromethylation of O‐, S‐, N‐, C‐nucleophiles using difluoromethyltri(n‐butyl)ammonium chloride as a new difluorocarbene source[J]. Chin J Chem, 2011, 29: 2717-2721. DOI:10.1002/cjoc.201100325 |
| [31] |
Bayarmagnai B, Matheis C, Jouvin K, et al. Synthesis of difluoromethyl thioethers from difluoromethyl trimethylsilane and organothiocyanates generated in situ[J]. Angew Chem Int Ed, 2015, 127: 5845-5848. DOI:10.1002/ange.201500899 |
| [32] |
Zhu D, Gu Y, Lu L. N-Difluoromethylthiophthalimide: a shelf-stable, electrophilic reagent for difluoromethylthiolation[J]. J Am Chem Soc, 2015, 137: 10547-10557. DOI:10.1021/jacs.5b03170 |
| [33] |
Zhu D, Hong X, Li D. A two-step, one-pot, and multigram-scale synthesis of N-difluoromethylthiophthalimide[J]. Org Process Res Dev, 2017, 21: 1383-1387. DOI:10.1021/acs.oprd.7b00203 |
| [34] |
Xiong HY, Pannecoucke X, Besset T. Recent advances in the synthesis of SCF2H- and SCF2FG containing molecules[J]. Chem Eur J, 2016, 22: 1-17. DOI:10.1002/chem.201504553 |
| [35] |
Ni C, Hu J. Recent advances in the synthetic application of difluorocarbene[J]. Synthesis, 2014, 46: 842-863. DOI:10.1055/s-0033-1340856 |
| [36] |
Sasson R, Hagooly A, Rozen S. Novel method for incorporating the CHF2 group into organic molecules using BrF3[J]. Org Lett, 2003, 5: 769. DOI:10.1021/ol034051n |
| [37] |
Mykhailiuk PK. New life for diazoacetonitrile (N2CHCN): in situ generation and practical synthesis of CN‐pyrazoles[J]. Eur J Org Chem, 2015, 33: 7235-7239. |
| [38] |
Duan YY, Lin JH, Xiao JC, et al. Difluoromethylcarbene for iron-catalyzed cyclopropanation[J]. Chem Commun, 2017, 53: 3870-3873. DOI:10.1039/C7CC01636K |
| [39] |
Hayashi H, Sonoda H, Fukumura K. 2, 2-Difluoro-1, 3-dimethylimidazolidine (DFI). A new fluorinating agent[J]. Chem Commun (Camb), 2002, 15: 1618-1619. |
| [40] |
Geri JB, Wade Wolfe MM, Szymczak NK. The difluoromethyl group as a masked nucleophile: a lewis acid/base approach[J]. J Am Chem Soc, 2018, 140: 9404-9408. DOI:10.1021/jacs.8b06093 |
| [41] |
Umemoto T, Singh RP, Xu Y, et al. Discovery of 4-tert-butyl-2, 6-dimethylphenylsulfur trifluoride as a deoxofluorinating agent with high thermal stability as well as unusual resistance to aqueous hydrolysis, and its diverse fluorination capabilities including deoxofluoro-arylsulfinylation with high stereoselectivity[J]. J Am Chem Soc, 2010, 132: 18199-18205. DOI:10.1021/ja106343h |
| [42] |
Lal GS, Pez GP, Pesaresi RJ, et al. Bis(2-methoxyethyl)aminosulfur trifluoride: a new broad-spectrum deoxofluorinating agent with enhanced thermal stability[J]. J Org Chem, 1999, 64: 7048-7054. DOI:10.1021/jo990566+ |
| [43] |
Umemoto T, Singh RP. Arylsulfur chlorotetrafluorides as useful fluorinating agents: deoxo- and dethioxo-fluorinations[J]. J Fluor Chem, 2012, 140: 17-27. DOI:10.1016/j.jfluchem.2012.03.008 |
| [44] |
Singh RP, Chakraborty D, Shreeve JM. Nucleophilic trifluoromethylation and difluorination of substituted aromatic aldehydes with Ruppert's and Deoxofluor reagents[J]. J Fluor Chem, 2001, 111: 153-160. DOI:10.1016/S0022-1139(01)00447-X |
| [45] |
Xu L, Vicic DA. Direct difluoromethylation of aryl halides via base metal catalysis at room temperature[J]. J Am Chem Soc, 2016, 138: 2536-2539. DOI:10.1021/jacs.6b00053 |
| [46] |
Tung TT, Christenson SB, Nielsen J. Difluoroacetic acid as a new reagent for direct C-H difluoromethylation of heteroaromatic compounds[J]. Chem Eur J, 2017, 23: 18125-18128. DOI:10.1002/chem.201704261 |
| [47] |
Mykhailiuk PK. In situ generation of difluoromethyl diazomethane for[J]. Angew Chem Int Ed, 2015, 54: 6558-6561. DOI:10.1002/anie.201501529 |
| [48] |
Khutorianskyi A, Chalyk B, Borysko P. Difluoromethyl nitrile oxide (CF2HCNO): a neglected chemical reagent[J]. Eur J Org Chem, 2017, 2017: 3935-3940. DOI:10.1002/ejoc.201700764 |
| [49] |
Li Z, Cui Z, Liu ZQ. Copper- and iron-catalyzed decarboxylative tri- and difluoromethylation of α, β-unsaturated carboxylic acids with CF3SO2Na and (CF2HSO2)2Zn via a radical process[J]. Org Lett, 2013, 15: 406-409. DOI:10.1021/ol3034059 |
| [50] |
Arai Y, Tomita R, Ando G. Oxydifluoromethylation of alkenes by photoredox catalysis: Simple synthesis of CF2H-containing alcohols[J]. Chem Eur J, 2016, 22: 1262. DOI:10.1002/chem.201504838 |
| [51] |
Shen X, Liu Q, Luo T, et al. Nucleophilic difluoromethylation of epoxides with PhSO(NTBS)CF2H by a preorganization strategy[J]. Chem Eur J, 2014, 20: 1-7. DOI:10.1002/chem.201390210 |
| [52] |
Guo SH, Zhang XL, Pan GF. Synthesis of difluoromethylthioesters from aldehydes[J]. Angew Chem Int Ed, 2018, 57: 1663-1667. DOI:10.1002/anie.201710731 |
| [53] |
Li Z, Dong J, Yuan Z. One-pot synthesis of 3-difluoromethyl benzoxazole-2-thiones[J]. Org Lett, 2018, 20: 6407-6410. DOI:10.1021/acs.orglett.8b02713 |
| [54] |
Huang Y, Lin Z, Chen Y, et al. Transition-metal-free N-difluoromethylation of hydrazones with TMSCF2Br as the difluoromethylation reagent[J]. Org Chem Front, 2019, 6: 2462-2466. DOI:10.1039/C9QO00506D |
| [55] |
Prakash GKS, Krishnamoorthy S, Ganesh SK, et al. N-Difluoromethylation of imidazoles and benzimidazoles using the Ruppert-Prakash reagent under neutral conditions[J]. Org Lett, 2014, 16: 54-57. DOI:10.1021/ol403007j |
| [56] |
Mao T, Zhao L, Huang Y. N-Difluoromethylation of imidazoles and pyrazoles using BrCF2PO(OEt)2 under mild condition[J]. Tetrahedron Lett, 2018, 59: 2752-2754. DOI:10.1016/j.tetlet.2018.06.003 |
| [57] |
Polley A, Bairy G, Das P. Triple mode of alkylation with ethyl bromodifluoroacetate: N, or O-difluoromethylation, N-ethylation and S-(ethoxycarbonyl)difluoromethylation[J]. Adv Synth Catal, 2018, 360: 4161-4167. DOI:10.1002/adsc.201800824 |
| [58] |
Zafrani Y, Saphier S, Gershonov E. Utilizing the CF2H moiety as a H-bond-donating group in drug discovery[J]. Fut Med Chem, 2020, 12: 361-365. DOI:10.4155/fmc-2019-0309 |
| [59] |
Jeffries B, Wang Z, Felstead HR, et al. A systematic investigation of lipophilicity modulation by aliphatic fluorination motifs[J]. J Med Chem, 2020, 63: 1002-1031. DOI:10.1021/acs.jmedchem.9b01172 |