 2021, Vol. 56
2021, Vol. 56

1977年FDA批准阿斯利康公司药物他莫昔芬(1, tamoxifene) 在美国上市, 治疗绝经期雌激素呈阳性的乳腺癌患者, 开启了科学意义的选择性雌受体调节剂(SERM) 的治疗领域, 标志着雌受体药物研究达到新的水平。由于以拮抗为主作用的他莫昔芬也具有一定程度的激动剂作用, 这种激动作用对于完全拮抗剂而言, 是脱靶作用。礼来公司的研发目标是雌受体的完全拮抗剂。
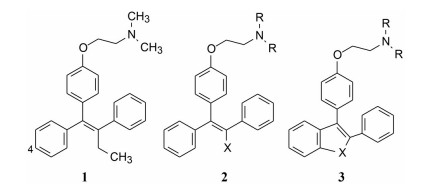
1.1 已有SERM的结构类型20世纪80年代国际上研发SERM者很多, 结构类型主要有两类: 三苯乙烯类(2, X为烷基、Cl或NO2等) 和环状等排物(3, X为CH2、NH、S或O), 两种骨架都含有烷胺乙氧基链, 药效团特征相似。
|
他莫昔芬是前药, 体内经代谢活化生成4-羟基他莫昔芬呈现拮抗作用。体外实验发现, 4-羟基他莫昔芬可发生异构化, 使本来的反式体异构化成顺-反体平衡态, 顺式体无效, 这或许是不研发4-羟基他莫昔芬的原因之一(Ruenitz PC, Bagley RJ, Mokler CM. Estrogenic and antiestrogenic activity of monophenolic analogues of tamoxifen, (Z)-2-[p-(1, 2-diphenyl-1-butenyl)phenoxy]-N, N-dimethylethylamine. J Med Chem, 1982, 25: 1056-1060), 不过在体内4-羟基他莫昔芬未发现有异构化的证据(Robertson DW, Katzenellenbogen JA, Hayes JR, et al. Antiestrogen basicity-activity relationships: a comparison of the estrogen receptor binding and antiuterotrophic potencies of several analogs of (Z)-1, 2-diphenyl-1-[4-[2-(dimethylamino)ethoxy]phenyl]-1-butene (Tamoxifen, Nolvadex) having altered basicity. J Med Chem, 1982, 25: 167-171)。而含有羟基的环状化合物骨架稳固, 不会发生异构化, 因而可以预制羟基于结构中。
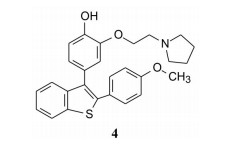
BMS公司曾研究了2, 3-二苯基苯并噻吩为骨架的化合物, 环上分别含有碱性侧链和羟基或甲氧基, 例如化合物4具有很强的抗生育活性, 是雌受体拮抗剂(Crenshaw RR, Jeffries AT, Luk GM, et al. Potential antifertility agents. 1. substituted diary1 derivatives of benzo[b]thiophenes, benzo[b]furans, lH-2-benzothiapyrans, and 2H-1-benzothiapyrans. J Med Chem, 1971, 14: 1185-1190), 但未获成功。
|
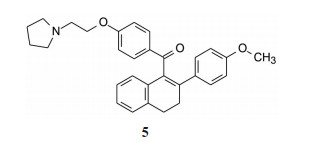
礼来公司研发的SERM也是环状化合物, 最初是以二氢萘为骨架, 并在骨架与连有碱性侧链的苯环之间插入一个羰基。加入的原委不得而知, 估计是赋予化合物的结构新颖性以获得专利保护, 而且化学合成也比较容易(付氏反应)。这类化合物仍保持抗雌激素作用, 代表性的是曲沃昔芬(5, trioxifene), 为雌受体ERα强效抑制剂, 抗肿瘤活性强于他莫昔芬, 进行了临床研究, 但未能上市(Jones CD, Suarez T, Massey EH, et al. Synthesis and antiestrogenic activity of [3, 4-dihydro-2-(4-methoxypheny1)-1-naphthalenyl] [4-[2-(1-pyrrolidiny1)ethoxyl-phenyllmethanone, methanesulfonic acid salt. J Med Chem, 1979, 22: 962-966)。
|
用幼年雌性大鼠评价化合物促进子宫生长或(和) 抗子宫生长, 作为雌受体激动或(和) 拮抗作用的指标。每日给药一次, 方法是只给受试物以评价促子宫生长作用; 给受试物加一定量雌二醇, 评价抗雌激素作用。第4日牺牲动物, 称量子宫重量(湿重)。对于活性高的化合物还测定剂量-效应的相关性。
化合物的抗肿瘤活性是用致癌剂DMBA引起大鼠乳腺肿瘤的模型评价抑制作用。用55日龄大鼠一次灌胃7, 12-二甲基苯并蒽(DMBA) 20 mg, 6周后乳腺出现肿瘤, 灌胃受试化合物10 mg·kg-1, 每日2次, 每周测量肿瘤面积, 记录最大和最小的数值。
化合物与雌受体的结合作用是用MCF-7细胞溶解液(含有雌激素受体) 与3H-雌二醇的结合被受试物置换的相对活性强度(RBA) 来表示。
化合物抗乳腺癌细胞活性是用1×10-11 mol·L-1雌二醇刺激MCF-7的增殖为模型, 测定不同浓度的受试物抑制细胞生长作用, 计算半数抑制浓度(IC50)。
体内评价化合物对组织特异性雌受体的激动作用是用75日龄去卵巢大鼠(OVX) 为模型, 评价的终末点包括: 测定子宫重量、测定子宫嗜酸性粒细胞过氧化酶(EPO) 活性、测定血清中胆固醇浓度、测定大鼠的骨密度。测定胆固醇水平和骨密度的原因, 是他莫昔芬的临床发现有保持骨密度和降低血清胆固醇的作用, 提示雌受体有不同的组织分布, 拮抗剂的特异性组织分布可能成为不同治疗领域的药物。
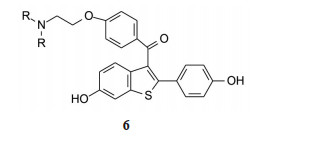
2.2 先导物的设计为了研发新结构类型的选择性拮抗剂, 礼来公司设计的先导物是将曲沃昔芬的二氢萘片段用6-羟基苯并噻吩替换(6), 拼合了曲沃昔芬含有羰基的结构和BMS研发的苯并噻吩母核, 在母核的2位连接4'-羟基苯, 3位连接带有碱性侧链的苯甲酰基。由于母核的两个芳环上预构了羟基, 有利于与受体的结合而无需体内代谢活化, 而且, 环状结构不会发生异构化。
|
将母核固定为4-羟基-2-(4'-羟苯基)-3-(4-取代的苯甲酰基)-6-羟基苯并噻吩, 只变换碱性侧链氮上烷基, 考察对雌鼠促子宫生长(受体激动作用)和抗子宫生长(受体拮抗作用) 的影响, 结果列于表 1, 构效关系表明: ①末端为四氢吡咯(7)、哌啶(8) 和氮杂环庚烷(9) 时, 随剂量提高, 抗子宫生长作用指数级增加, 而促子宫增长作用没有变化, 提示含环状胺基侧链的化合物为雌受体拮抗剂; ②末端为链烷胺基(10~12) 时, 子宫增重随剂量提高而增加, 提示有雌激素样作用, 因而抑制了部分拮抗作用(子宫生长减弱), 在高剂量(100~1 000 μg) 对抗雌二醇作用尤甚。
| Table 1 The structures and activity of compounds that change basic side chains |

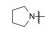
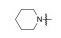
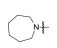
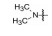
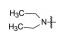
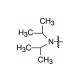
由此推论环烷与链烷的变化对激动-拮抗有重要区分, 完全拮抗剂的碱性侧链宜为环状胺基。
化合物8在高剂量1 000 μg时达到最大的抑制雌二醇作用(> 90%), 显著强于他莫昔芬活性, 化合物代号为LY15675, 拟作为候选化合物(Jones CD, Jevnikar MG, Pike AJ, et al. Antiestrogens. 2. Structure-activity studies in a series of 3-aroyl-2-arylbenzo[b] thiophene derivatives leading to [6-hydroxy-2-(4-hydroxyphenyl)benzo[b]thien-3-yl] [4-[2-(1-piperidiny1)ethoxyl-phenyllmethanone hydrochloride (LY 156758), a remarkably effective estrogen antagonist with only minimal intrinsic estrogenicity. J Med Chem, 1984, 27: 1057-1066)。
2.3.2 变换6-羟基和4'-羟基化合物8是碱性侧链中活性最高的化合物, 下一步是固定该侧链不变, 同时变换6-OH和4'-OH, 考察这两个位置的基团对活性的影响, 合成的化合物及其活性列于表 2。分析构效关系如下: ①当化合物8的6-OH和4'-OH都被氢(13) 或其中一个被氢(18和38) 置换, 受体结合作用分别降低 > 100、100或5倍, 抑制癌细胞的活性也下降, 提示环上羟基是重要基团。②化合物8的6位羟基移至7、4或5位(25~27), 与受体结合和抑制癌细胞活性都明显减弱, 说明羟基位置的重要性。8的6-OH相当于雌二醇的3-OH, 雌二醇的3-羟基的邻位存在基团因位阻效应而降低活性, 同样化合物8在5或7位连接基团(如29、32~34) 也使活性降低, 然而4'-OH的邻位效应不显著(53~56), 提示6-OH对活性贡献大于4'-OH, 这与雌二醇的3-OH的重要性大于17β-OH相一致(Heahnel R, Twaddle E, Ratajczek T. Specificity of the estrogen receptor of human uterus. J Steroid Biochem, 1973, 4: 21-31)。③化合物8的4'-OH被卤素取代(20、24) 时, 受体结合与细胞抑制作用都减弱。羟基移至2'或3'位(39、40) 降低活性。④ 4'-OH被甲基取代(41) 或引入甲基(51) 仍然保持活性, 但增大烷基使活性下降, 如4'-n-Bu (45)。
| Table 2 The activity of compounds with different 6-, 4'-dihydroxy. *Dose required to give 50% inhibition of a maximally effective (1×10-11) dose of 17β-estradiol |
综上, 6-OH和4'-OH是两个优选的取代基团, 变换或失去或增加位阻, 都使活性降低。
2.3.3 变换2位苯基确定6-羟基苯并噻吩是优选的结构片段后, 下一步是考察2位苯环是否有优化空间, 为此, 合成了2-萘环、-噻吩、-烷基、-环烷基、-苄基和吡啶等, 化合物活性列于表 3。结果表明, 与雌受体结合和抑制癌细胞作用大都与8相近, 但2-萘基(75) 和吡啶化合物(86、87) 活性很弱, 可能与不适宜的体积或极性有关。
| Table 3 Activity of compounds with 2-phenyl change. *Dose required to give 50% inhibition of a maximally effective (1×10-11) dose of 17β-estradiol |

雌受体具有多种生物学活性, 他莫昔芬是以雌受体拮抗剂目标研发的, 其也呈现激动活性(部分拮抗剂), 但对骨骼和心血管系统呈现雌激素的激动作用, 研究表明某些SERM的组织特异性可对子宫和乳腺产生拮抗作用, 而对骨组织和心血管则为雌激素样激动作用。所以, 化合物对子宫的促(或抗) 生长作用不能推论对其他组织生物活性, 因为化合物的组织特异性难以预料。
SERM对子宫的促红细胞生成素(EPO, 调控红细胞生长的一种糖蛋白) 活性作为化合物对雌受体的作用指标。他莫昔芬和炔雌醇(后者是激动剂) 使子宫增殖以及EPO提高, 而化合物8却没有此作用, 所以, 依据对EPO的作用可区分他莫昔芬样的部分拮抗作用和化合物8的完全拮抗作用。对心血管作用指标是用血清中胆固醇水平表征。
对上述化合物进一步评价对OVX (去卵巢大鼠) 的子宫增重作用, 相对于对照组的子宫EPO活性(Vmax), 以及降低血清胆固醇50%的剂量(ED50)。表 4列出了化合物的活性。
| Table 4 Effects of compounds on uterine growth, EPO activity and serum cholesterol levels in ovariectomized rats. *MED at which a significant (> 5-fold increase relative to OVX control and value of Vmax ≥ 10) increase in EPO activity was observed. Activity at the MED is expressed as Vmax |
表 4的构效关系提示: ①化合物8失去6-OH和(或) 4'-OH (化合物13、15、37) 或被甲醚化(15、18) 可强效降低血清胆固醇水平, 略增重子宫, 但不增高EPO活性, 这与8相似。联想表 1的受体结合数据, 推论这些化合物发生了羟基化或去甲基化的代谢活化。②羟基处于其他位置, 如25~27、39和40, 则降低对胆固醇的作用。③苯并噻吩环的4, 5或7位引入基团(如32和33) 降低体内活性。
4 评价骨保护作用绝经后妇女雌激素水平低, 引起骨质疏松而易发生骨折。基于雌受体的组织特异性, 对有代表性化合物评价了去卵巢大鼠的骨保护作用, 表 5列出了剂量与股骨远端骨密度的保护作用的关系。
| Table 5 The protective effect of the compound on the bone mineral density of the distal femur |
表 5中的数据提示这些代表性的化合物对缺失性激素引起的骨质疏松有保护作用, 显示出对骨组织中的雌受体具有激动作用。在量效关系上相关性较低, 是因为测定方法的变数较多的缘故。
5 候选物和他莫昔芬上市以8为核心的结构变换, 进行了广泛的构效关系探索, 8仍显示为雌受体的强效结合剂, 对某些组织呈现激动作用(如骨组织) 而对某些组织为拮抗剂, 对子宫未呈现作用, 显示仍为强效的SERM, 但与他莫昔芬不同。虽然口服生物利用度较低, 在肠中发生II相代谢(葡萄糖醛酸苷化), 仍确定候选物, 经三期临床试验, 美国FDA批准了对绝经期妇女的骨质疏松预防(1997年) 和治疗(1999年) 的适应症(Grese TA, Cho S, Finley AG, et al. Structure-activity relationships of selective estrogen receptor modulators: modifications to the 2-arylbenzothiophene core of raloxifene. J Med Chem, 1997, 40: 146 167)。
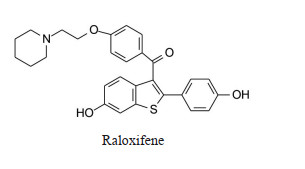
|
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和疗效评价等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
雌激素有多重生理功能, 雌激素受体的组织分布具有特异性, 因而选择性雌激素受体调节剂(SERM) 可研发不同领域的治疗药物。雷洛昔芬是雌激素受体的强效结合剂, 兼有激动和拮抗双重作用, 临床应用的主要是对骨组织的雌激素受体激动作用, 区别于首创药他莫昔芬是治疗ER呈阳性的乳腺癌药物。研发者对化学结构做广泛改造的同时, 评价了多种体内外药理指标, 在受体分布的特异性和化合物的拮抗-激动的互动印证中, 成功地研发出雷洛昔芬。
(编者按)