 2021, Vol. 56
2021, Vol. 56

新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
他莫昔芬是首创的选择性雌激素受体调节剂(SERM), 治疗雌激素受体呈阳性的乳腺癌。他莫昔芬的研发是ICI公司(今阿斯利康) 借鉴Merrell公司研发非甾体避孕药的结果, 坚持研究近20年而成功的重磅药物。研发中结构改造和优化没有特别的难度和复杂过程, 其曲折与艰巨性在于改换适应症, 确定作用靶标以及乳腺癌患者的适用群。药理作用在表型-靶标-概念验证的转化研究中, 显示基础研究和临床实践互动的重要性。他莫昔芬的上市开辟了“昔芬”类药物治疗领域, 跟随创制的药物治疗乳腺癌和骨质疏松等。此外, 在一定程度上还带动了选择性雌受体降解剂(selective estrogen receptor degraders, SERD) 的研制, 如氟维司群的上市成为小分子药物研制蛋白降解剂的始作俑者。
(编者按)
他莫昔芬是第一个上市的选择性雌激素受体调节剂(SERM), 1973年在英国上市, 1977年美国FDA批准上市, 间隔的4年, 是确证作用靶标和适应证的时日, 定为治疗雌受体呈阳性的乳腺癌患者的药物。从1958年启动研究, 到确定为科学意义上的乳腺癌治疗药, 历时20年。
他莫昔芬作为首创性药物, 研制过程有以下特点: 以研究避孕药开始, 最终研制出抗乳腺癌新药; 由对动物的表型评价, 归结到抑制雌受体靶标; 在表型-靶标-概念验证的转化研究中, 显示了新药研发中基础研究和临床实践的互动关系; 他莫昔芬的上市, 开辟了选择性雌受体调节剂(SERM)“昔芬”类药物治疗领域; 也引领了研制作用于核受体家族药物的路径, 对细胞生物学和药物创制具有重大意义。
1.1 生理表型研究促进靶标发现早在19世纪末叶临床实践表明, 绝经前患乳腺癌患者可通过切除卵巢得到缓解, 后来还发现大约三分之一的患者可通过这种方法治疗。进而采取的策略是阻断乳腺中的雌激素功能来治疗或预防乳腺癌的发生。
另一研究路径是在阐明生殖内分泌生理学中, 通过合成评价化合物的抗雌激素作用, 于20世纪60年代发现了雌激素受体(Jensen EV, Jacobson HI. Basic guides to the mechanism of estrogenaction. Rec Prog Hormone Res, 1962, 18: 387-414), 为研发治疗依赖于雌激素的乳腺癌提供了分子依据和准备了治疗药物靶标。
最初针对雌激素的药物研究都是以口服避孕药为目的, 但都没有成功。直到转向与雌激素受体相关的疾病, 在治疗癌症和骨质疏松获得了成功。失之东隅, 收之桑榆。
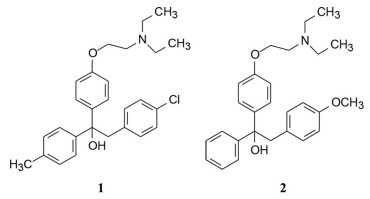
1.2 非甾体类雌激素拮抗剂20世纪50年代Merrell公司研发的降胆固醇药物曲帕拉醇(1, triparanol), 体内具有抑制胆固醇合成的作用, 作用机制是阻止生物合成中的最后环节, 链甾醇(desmosterol) 转变成胆固醇, 然而由于链甾醇在体内的蓄积, 引起白内障发生, 从而停止了曲帕拉醇的临床应用(Takahashi MP, Kimura T, Yanagihara T, et al. Calcium increase in mouse skeletal muscles by triparanol: a drug to induce myotonic dystrophy-like clinical manifestations. Neurosci Lett, 1999, 272: 87-90)。
Merrell公司对曲帕拉醇作结构改造, 化合物2 (MER25) 失去了降胆固醇活性。但在随机筛选对小鼠生殖器官的作用时, 意外地发现可对抗雌二醇的作用, 使小鼠子宫增重, 并证明化合物2对交配后的小鼠有避孕效果。然而化合物2临床试验表明避孕效果差, 后来用于治疗子宫内膜和乳腺癌, 效果仍然不佳, 而且还显示对中枢神经系统的不良作用。虽然化合物2没有成为药物, 却开启了非甾体化合物抗雌激素的研究(Kraft RO. Triparanol in the treatment of disseminated mammary carcinoma. Cancer Chemother Rep, 1962, 25: 113-115)。
|
与此同时, 其他公司如ICI (现为阿斯利康)、Upjohn和礼来等也在研发非甾体类雌受体调节剂, 但没有借鉴化合物2的结构。
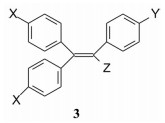
2 先导物的发现化合物2的抗雌激素活性较弱, 但作用持续时间长。其实, 早在1953年Merrell公司就研究三苯乙烯类化合物(3), 证明具有较弱但持久的雌激素作用, 结构中没有胺烷氧基侧链, 未呈现抗雌激素作用(Shelten RS, Van Campen MG Jr, Meisner DS, et al. Synthetic estrogens. halotriphenylethylene derivatives. J Am Chem Soc, 1953, 75: 5491-5495)。
|
Palopoli等将化合物2的中心碳原子的羟基经消除H2O, 合成了以氯代三苯乙烯为骨架的化合物, 合成了含有不同胺烷基链取代的化合物, 列于表 1中。这些化合物皮下注射雌性幼鼠, 测定化合物对性器官生长的影响, 与对照组的重量比较, 作为表征抑制促性腺激素的活性(Palopoli FP, Feil VJ, Allen RE, et al. Substituted aminoalkoxytriarylhaloethylenes. J Med Chem, 1967, 10: 84-86)。虽然没有报道这些化合物的详细数据, 但活性最强的化合物4, 对雌性大鼠每日以0.1 mg·kg-1低剂量给药, 卵巢可达到50% (重量) 的抑制率。抑制动物促性腺激素实验表明, 4兼有雌激素和抗雌激素双重活性(Holtkamp DE, Greslin JG, Root CA, et al. Gonadotrophin inhibiting and antifecundity effect of chloramiphine. Proc Soc Exp Biol Med, 1960, 105: 197-201)。同样在低剂量下对幼年雄鼠前列腺有增重作用。这些实验说明, 在低剂量下4对促性腺激素有刺激作用。
| Table 1 Structural transformation of halogenated tristyrene compounds (No activity data in literature) |
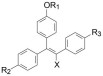
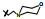
化合物4定名为氯米芬(clomiphene) 进入临床试验(20世纪60年代临床试验的门槛低), 发现可诱导妇女排卵, 而且没有呈现事后避孕作用, 表明氯米芬对人体也有双重活性: 雌激素作用和抗雌激素作用。更重要的是发现氯米芬对乳腺癌有治疗作用(Herbst AL, Griffiths CT, Kstner RW. Clomiphene citrate (NSC-35770) in disseminated mammary carcinoma. Cancer Chemother Rep, 1964, 43: 39-41)。临床用的氯米芬样品是顺反异构体的混合物, 20世纪70年代仍以混合物形式治疗妇女乳腺癌(Hecker E, Vegh I, Levy CM, et al. Clinical trial of clomiphene in advanced breast. Eur J Cancer, 1974, 10: 747-749)。
将氯米芬混合物分离成单体, 即反式体恩氯米芬(4E, enclomifene) 和顺式体珠氯米芬(4Z, zuclomifene), 动物实验表明4E对雌受体呈现拮抗作用, 4Z具有激动剂作用(Palopoli FP, Feil VJ, Allen RE, et al. Substituted aminoalkoxytriarylhaloethylenes. J Med Chem, 1967, 10: 84-86)。
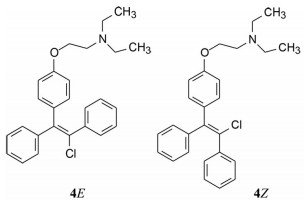
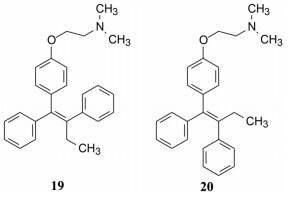
3 他莫昔芬的发现 3.1 新结构骨架的顺反异构体的活性ICI公司的内分泌专家Walepole自20世纪50年代研究激素类药物, 他与同事Harper和Rechardson得知Merrell公司停止了曲帕拉醇临床试验, 试图合成三苯乙烯化合物以去除不良反应(Harper MJ, Richardson DN, Walepole AL. Alkene derivative. GB patent 1013907A), 其中化合物19和20为含有胺烷氧基侧链的三苯乙烯衍生物, 分别为反式(无取代苯环处于双键异侧) 和顺式(苯环处于双键同侧), 二者互为立体异构。如同4E和4Z的活性差异一样, 19 (反式体) 对雌性大鼠显示抗雌激素作用, 可以抑制因注射雌激素引起雌鼠阴道角化和子宫增重作用, 且终止大鼠排卵和早期妊娠, 但对小鼠则为雌激素活性, 提示种属差异引起活性翻转; 顺式体20对大鼠或小鼠都具有雌激素活性。表 2列出了化合物19和20对大鼠的雌激素作用, 各项指标都显示19具有抗雌激素作用, 20有雌激素样作用(Harper MJ, Walepole AL. Contrasting endocrine activities of cis and trans isomers in a series of substituted triphenylethylenes. Nature, 1966, 212: 87; Bedford GR, Richardson DN. Preparation and identification of cis and trans isomers of a substitutedtriarylethylene. Nature, 1966, 212: 733-734)。
|
|
| Table 2 The estrogen effect of compounds 19 and 20 on rat |
在公开发表的文章和专利中没有讨论确定化合物19为候选化合物的更多依据, 例如碱性侧链的结构与活性的关系。不过, 当初在ICI参与项目的化学家Richardson后来合成了不同的碱性侧链, 考察碱性对活性的关系。表 3列出的化合物的结构、pKa及其活性。
| Table 3 In vitro activity and pKa of compounds with different side chains. *Estradiol=100% |
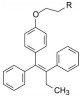
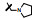

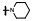
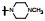

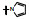
表 3的数据表明这些化合物与雌受体有一定的结合作用, 但不强, 尤其是和27~29的活性很弱, 推测与较弱的碱性相关联。然而, 如同后面讨论19的代谢作用, 由于这些化合物苯环上没有羟基, 受体结合作用都较弱, 掩盖了碱性和侧链结构的变化对活性影响的差异。
这些化合物给药雌性幼鼠3天, 测定子宫的重量, 表明22~27的抗雌激素活性(对抗外加雌激素的作用) 与19相当(Robertson DW, Katzenellenbogen JA, Hayes JR, et al. Antiestrogen basicity-activity relationships: acomparison of the estrogen receptor binding and antiuterotrophic potencies of several analogues of (2)-1, 2-diphenyl-1-[4-[2-(dimethy1amino)ethoxy]phenyl]-1-butene (tamoxifen, nolvadex) having altered basicity. J Med Chem, 1982, 25: 167-171)。
3.3 化合物19的代谢活化由于化合物19对小鼠的作用与大鼠不同, 因而Walepole担心种属差异对人的避孕效果, 没有做深入研究。幸亏他对肿瘤治疗尤感兴趣, 在专利中申请并获得了这类化合物控制与激素相关肿瘤的权利要求, 这对ICI公司研发他莫昔芬是至关重要的。
化合物19定名为他莫昔芬(tamoxifen), 1972年ICI进行临床试验, 并于1973年批准在英国上市, 用于治疗多种疾病: 抑制排卵, 治疗乳腺癌、月经过多等, 但效果都欠佳。尤其对乳腺癌的治疗没有得到普遍的临床认可(主要应用于生殖内分泌的抗雌激素方面), 每年销售额只有几十万英镑。
确定他莫昔芬治疗乳腺癌首创地位归因于基础研究, 一是发现并确定了雌激素受体靶标, 另一是用致癌剂二甲基苯并蒽(DMBA) 诱导大鼠乳腺癌模型。也由于专利保护了ICI公司的他莫昔芬的抗肿瘤用途, 促进了深入研究其作用机制。
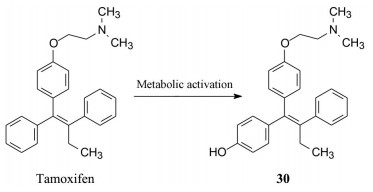
Jordan于1972~1974年在Jensen (雌受体发现者) 指导下, 深入研究了他莫昔芬体外抗雌激素作用和大鼠体内抗乳腺癌作用(JordanVC. Effect of tamoxifen (ICI-46474) on initioation and growth of DMBA-induced rat mammary carcinoma. Eur J Cancer, 1976, 12: 419-424), 以及二者之间的关系, 发现他莫昔芬体外抑制雌受体的活性很低, 而对大鼠子宫的抗生育作用很强, 提示可能是体内代谢活化的缘故, 从而发现了代谢产物4-羟基他莫昔芬(30), 体外对雌受体呈现很强的抑制活性, 证明了体内抑制大鼠乳腺癌的生长与抑制乳腺癌的雌激素受体相关, 奠定了临床治疗的基础。进而临床证实他莫昔芬对乳腺癌雌受体呈阳性的患者是有效的, 对阴性患者没有作用。美国FDA与1977年批准他莫昔芬在美国上市治疗雌受体呈阳性的乳腺癌患者(Jordan VC. Tamoxifen: a most unlikely pioneering medicin. Nat Rev Drug Disc, 2003, 2: 205-213)。
|
所以, 他莫昔芬是前药, 在体内肝脏CYP3A4、2C9和2D6作用下, 生成4-羟基他莫昔芬(30), 后者未开发成药物, 是由于酚羟基在体内容易发生Ⅱ相代谢, 首过效应降低生物利用度。