 2021, Vol. 56
2021, Vol. 56



局部黏着斑激酶(focal adhesion kinase, FAK), 也称之为蛋白酪氨酸激酶2 (protein tyrosine kinase 2, PTK2), 是一种非受体细胞内酪氨酸激酶, 通过其激酶活性和支架功能在癌细胞的黏附、存活、代谢、增殖、迁移和肿瘤微环境中发挥着重要作用[1]。FAK的过表达及激活主要在原发性癌及转移性癌中被发现, 如原发性肝癌、结肠癌、卵巢癌、乳腺癌等多种不同人源癌细胞中均有FAK蛋白的过表达, 并表现出较差的临床治疗结果。FAK的作用机制及信号通路已得到广泛研究, 最近研究表明FAK是某些癌症中免疫应答的关键媒介, 抑制FAK的活性可能会触发免疫介导的肿瘤消失, 证明FAK是治疗癌症的有效潜在靶点[2]。鉴于在癌症细胞中FAK的过表达, 当前已有许多直接作用于FAK的小分子抑制剂, 且有少部分已进入临床研究[3]。尽管靶向FAK的小分子抑制剂在临床前实验中取得令人兴奋的成绩, 临床试验也取得阶段性的成功, 但还远远未满足临床需要[4]。由于传统药物化学策略如小分子抑制剂只能抑制FAK的激酶依赖性的生理活性而忽略了FAK非激酶依赖的支架角色功能, 其作用机制大大限制了靶向FAK的临床应用[5]。为了克服FAK激酶抑制剂的作用机制缺陷, 深入靶点FAK的认识, 验证靶点FAK的临床应用, 探讨支架功能在肿瘤发生发展中的作用, 迫切需要全新的策略来选择性靶向。
蛋白水解靶向嵌合体(proteolysis targeting chimera, PROTAC) 技术是一种新兴的药物研发策略[6, 7]。特别是在2019年2个新型可口服小分子PROTAC ARV-110 (NCT03888612) 和ARV-471 (NCT04072952) 进入临床Ⅰ期试验并取得令人满意的疗效以及安全性, 极大地鼓舞了来自学术界及制药工业界的科研人员研发热情[8]。PROTAC是由三部分组成的异源双功能化合物, 分别是用于特异性结合靶蛋白的"弹头" (warhead)、招募E3泛素连接酶的E3泛素连接酶配体(E3 ligand) 和连接两者的连接链(linker)[9]。PROTAC靶向降解靶蛋白的作用机制是通过嵌合体分子将靶蛋白与E3泛素连接酶"拉近"到适当距离, 使得本不被特异性泛素化的蛋白得到泛素化标记, 最终通过蛋白酶体系统降解蛋白使其丧失生物学活性。与传统抑制剂只能抑制某些多功能蛋白的单一信号通路相比, PROTAC分子可以诱导靶蛋白降解从而影响该蛋白的所有功能, 从而减少反馈机制, 在某种程度上可以克服耐药以及减小耐药的发生几率[10]。从PROTAC的作用机制上看, 比起高亲和力的传统小分子抑制剂, 嵌合体分子对靶蛋白的亲和力要求并不苛刻, 只需微小的结合力便能诱导靶蛋白泛素化并被随后的泛素蛋白酶系统降解[11]。另一方面, PROTAC小分子可以发挥亚化学剂量的催化作用, 解离后的PROTAC分子可以进入下一个循环, 从而可以大大减少给药剂量以及降低因高剂量带来的毒副作用(图 1)。综合PROTAC的诸多优势, PROTAC不仅促进了小分子药物的研发, 更预示着第四次制药革命的到来[12]。

|
Figure 1 The mechanism of degradation of PROTAC |
PROTAC的独特作用机制能靶向降解FAK蛋白, 从而消除FAK的支架功能, 极大吸引了科研人员的兴趣[13]。当前已有多个基于PROTAC技术研发的靶向FAK小分子降解剂被报道[14]。本文简述了FAK蛋白、信号通路及小分子抑制剂, 系统综述了基于PROTAC技术靶向降解FAK蛋白的最新研究进展, 最后总结并展望了基于PROTAC技术靶向降解FAK蛋白的发展前景。
1 FAK的结构及信号通路FAK结构最早于1992年被Schaller等[15]从鸡胚成纤维细胞中分离并克隆出来。FAK隶属于酪氨酸激酶-2基因编码的非受体型酪氨酸激酶, 不含常见的SH2与SH3结构域, 是较为纯粹的胞质酪氨酸激酶, 主要依赖其细胞整合素发挥作用。FAK本身虽非原癌基因, 但易受到原癌基因、整合素、内皮素、升压素等的影响发生催化底物蛋白酪氨酸残基的磷酸化作用, 进而调控细胞的增殖、分化与生长[16]。编码FAK的cDNA长度为3 791 bp, 位于第8号染色体的8q24处, 可以编码1 052个氨基酸, 相对分子质量为125。FAK是一种具有多功能结构域的信号蛋白质, 主要有三部分组成: ①约有370个氨基酸组成的三叶草型的FERM和Y397磷酸化位点的N端; ②由150个氨基酸组成的FAT, 含大量脯氨酸聚集的和Y925的磷酸化位点的C端; ③包含Tyr576和Tyr577两个磷酸化位点的中心激酶结构域三部分[17] (图 2)。三者的共同作用介导着FAK的非激酶依赖的信号通路[18]。FAK通过其支架功能结构域参与在质膜上形成大型信息复合物, 其很容易被生长因子和整合素磷酸化激活, 介导胞外信号传至胞内, 连接细胞外基质与细胞的相互作用[19, 20], 并介导多条信号转导通路, 如P13K-AKT通路、Ras-MAPK通路、GTP/STAT1通路等, 参与调控肿瘤细胞的生长、增殖、转移和侵袭等过程[21]。

|
Figure 2 Schema of the structure of FAK and FAK - related signaling pathways |
生物学研究表明, 编码FAK蛋白的基因本身并不属于原癌基因, 但FAK在肿瘤源中的核酸及蛋白的过度表达和其自身活性的升高, 都会很大程度上影响肿瘤的发生与发展。FAK能调控癌细胞的增殖、凋亡和迁移等过程[22]。
2.1 调控癌细胞的增殖与凋亡肿瘤的发生, 究其原因是癌细胞增殖与凋亡的平衡失常, 从而减缓癌细胞的凋亡, 加快癌细胞的增殖, 抑或是凋亡敏感性不足等[23]。在肿瘤细胞中, FAK的过表达能够抑制细胞的"失巢凋亡", 从而阻断细胞与其外基质脱离接触而诱发的细胞正常性死亡。此外, FAK可以通过结合胞质内的P53功能蛋白, 促进P53的降解, 抑制了P53的转录活性与其介导的凋亡作用, 从而促进癌细胞的生长和发育[24]。
2.2 调控癌细胞的转移与侵袭癌细胞的转移是体内癌细胞迁移的结果[25]。研究表明, FAK是多条重要信号通路的交汇站, 其信号通路和刺激因子的受体都可激活FAK磷酸化, 调节肿瘤细胞黏附与迁移。在其转移、侵袭等过程中, FAK能够促进癌细胞Src、Ras、Apexl、Rgnef的mRNA及蛋白质表达, 调控癌细胞生长转移等。最近研究表明, 抑制FAK表达能有效抑制胃癌、肝癌等细胞中的Src、Ras、Apexl、Rgnef表达[26]。
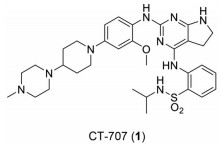
2.3 调控癌细胞通路1在大多肿瘤细胞中, FAK能激活P13K-AKT通路, 使其处于异常活化状态, AKT与mTOR皆处在高度磷酸化状态, 抑制FAK对该通路的活化, 会抑制癌细胞的增殖及恶化[27]。最新研究表明, 新型多靶点激酶抑制剂CT-707 (1) 正是通过靶向FAK以激活AKT相关通路, 从而克服对克唑替尼耐药的非小细胞肺癌的治疗[28]。
|
FAK-Ras-MAPK通路是FAK介导的主要转导信号通路之一[29]。Ras蛋白有两种状态, 当结合GTP时为活化状态, 反之当结合GDP为失活状态。MAPK作为胞内的一种蛋白激酶, 它的表达调节着细胞的分化生长等。激活的FAK通过激活Ras, 进而激活MAPK, 达到调节癌细胞的增殖, 分化与生长等作用, 进而密切联系了FAK与癌细胞之间的分化与生长的关系。
3 FAK小分子抑制剂当前已有较多FAK小分子抑制剂被报道, 尽管还未有直接作用于FAK的小分子抑制剂上市, 但其体外、体内、临床前和临床试验研究数据均表现出令人满意的结果。目前已经大量报道了通过不同作用机制, 靶向FAK不同结构域的小分子抑制剂, 主要可以分为: 别构位点抑制和ATP竞争性抑制的激酶抑制剂、FERM结构域抑制剂、FAT结构域抑制剂。
FAK的ATP竞争性抑制剂是所有FAK抑制剂中研究最多的类别, 当前进入临床研究的均属于该类抑制剂。现已有6个直接作用于FAK的ATP竞争性抑制剂处于临床Ⅰ/Ⅱ期研究(表 1、图 3)。
| Table 1 FAK inhibitors that have been reported in clinical trials |

|
Figure 3 Inhibitors of FAK advanced in clinical trials |
此外, 还有较多活性较好FAK的ATP竞争性抑制剂被报道, 结构如图 4。

|
Figure 4 Representative inhibitors of FAK |
近日, 中国药科大学孙海鹰教授[30]系统综述了FAK小分子抑制剂最新研究进展, 本文由于篇幅有限, 直接作用于FAK其他结构域的小分子抑制剂本文不再赘述。
4 FAK降解剂的最新研究进展由于FAK同时具有激酶依赖性和独特的非激酶依赖性的支架功能, 而传统的小分子抑制剂只能抑制其特定的激酶活性, 不能有效靶向其非激酶的支架功能, 加之当前鲜有文献报道关于FAK的支架功能在肿瘤发生发展中的作用, 因此迫切需要寻找一个新的策略来更为深入的探讨FAK靶点的可成药性及对该靶点的系统研究。PROTAC技术的出现可以弥补传统小分子抑制剂不可靶向支架功能的缺陷。因此, 基于PROTAC技术研发靶向降解FAK成为研究FAK支架功能的热点, 更为研发靶向FAK的小分子药物带来新策略。
4.1 基于CRBN靶向FAK的小分子PROTACsCereblon (CRBN) 是一种与肿瘤密切相关的蛋白, 是E3泛素连接酶中的一种连接酶复合物的底物识别构件, 这个底物识别构件名为: CUL4-RBX1-DDB1-CRBN。此识别构件可以促使泛素特异性结合到底物蛋白上, 因此需要降解的蛋白就可以被标记出来[31]。CRBN配体由于分子量小、成药性好、便于合成等优势以广泛用于PROTACs小分子的设计与合成[32]。
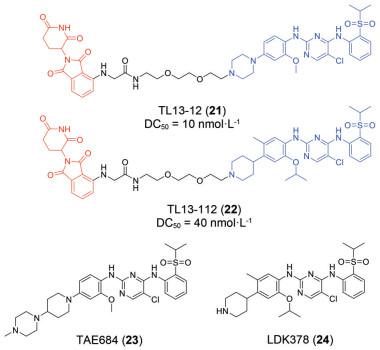
2018年Gray教授领衔的研究团队[33]首次报道了降解FAK的小分子PROTACs。降解剂TL13-12 (21) 和TL13-112 (22) 分别是基于间变性淋巴瘤激酶(ALK) 小分子抑制剂TAE684 (23) 和LDK378 (24) 所设计的。值得注意的是, 较ALK小分子抑制剂, 基于PROTAC研发的ALK降解剂药理活性得到明显提升。有趣的是, 作者研究之初旨在靶向降解ALK, 但所设计的PROTAC分子不仅可以降解ALK还可以降解FAK、Aurora A、FER和RPS6KA1, 表现出多靶点特性。推测其主要原因是PROTAC"弹头"本身具有激酶泛选择性。尽管当前已有相关文献报道通过PROTAC策略可以增加对靶蛋白的选择性, 但是对其增加选择性的作用机制仍不清楚, 受到"弹头"、连接链和E3泛素连接酶三部分的共同影响, 未来还需要在PROTAC提高选择性方面做更深入的研究。虽然降解FAK不是该研究的主要目的, 但是该研究证明了运用PROTAC降解FAK蛋白的可行性, 为后续降解FAK奠定了理论基础。
同年, Gray团队以ALK小分子抑制剂TAE684 (23) 为先导化合物通过结构修饰, 去掉了在选择性中起关键作用的2-苯胺上的2-甲氧基以降低其选择性, 合成了具有多激酶抑制活性功能的化合物TL13-87 (25)[34]。为了实现泛激酶降解活性, 研究人员引入了PROTAC策略, 用聚乙二醇连接链连接TL13-87 (25) 和CRBN配体来那度胺得到了具有同时降解包括FAK在内的28种激酶活性的小分子TL12-186 (26) 降解剂。
|
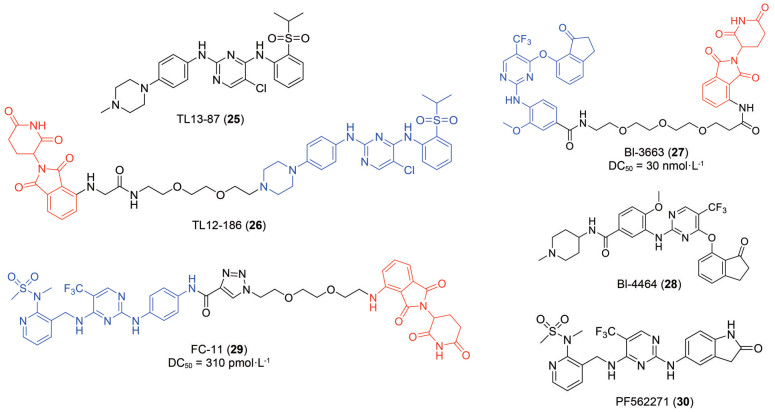
鉴于先前报道的诸多关于FAK的PROTAC选择性均较低, 为实现高选择性以降低毒副作用, 2019年Popow等[35]发现了具有高选择性PROTAC分子BI-3663 (27)。BI-3663 (27) 是由该团队自行研发具有高选择性的FAK抑制剂BI-4464 (28) 与CRBN配体泊马度胺通过含有3个单元的PEG连接链连接而成。降解实验表明, 11 HCC细胞系给予BI-3663 (27) 孵育18 h, DC50 = 30 nmol·L-1, 且大于80%的FAK蛋白被有效降解, 表现出较好的降解活性。尽管BI-3663 (27) 对FAK蛋白的降解具有较好活性, 但是其抗增殖活性较FAK小分子抑制并没有得到显著增强。因此, 本项研究质疑了FAK的支架功能在癌细胞增殖过程中的必要性, 但还需要更加深入的研究。总之, 本研究为研究FAK支架功能提供了有效的探针分子, 为后续研究奠定基础。
为实现高效、快速及选择性高的FAK降解剂, 2019年清华大学饶燏教授团队[36]基于CRBN配体建立了FAK小分子PROTACs化合物库。为了提高合成效率, 该团队通过"点击化学"引入三氮唑, 高效合成34个目标PROTACs分子, 其中FC-11 (29) 表现出较好的降解活性(DC50 = 310 pmol·L-1)。FC-11 (29) 的降解活性能够达到皮摩尔级别, 结合先前的文献报道, 推测可能是三氮唑的引入大大增强了FAK和E3泛素连接酶配体的蛋白-蛋白相互作用, 从而增强降解活性。此外, 该研究还系统讨论了连接链的长度, PROTAC与两配体的结合模式和两配体的空间趋向对降解FAK活性的影响。但是该影响可能存在较多偶然因素, 当前对PROTAC的理性药物设计还没有规律的认识, 未来还需要更广泛的研究, 形成统一的认识。
2020年, 饶燏教授团队[37]为了研究FAK的激酶依赖与其非激酶依赖的支架功能对雄性小鼠生殖系统的影响, 实施了连续给药13天的PROTAC小分子FC-11 (29) (20 mg·kg-1, BID)、抑制剂PF562271 (30) (10 mg·kg-1, BID) 和空白组的动物实验。实验结果表明, 13天给药后, 给药FC-11 (29) 的雄性小鼠在睾丸、附睾、精囊和包皮腺等器官中表现出显著的质量及体积的变小。此外, FC-11 (29) 处理的小鼠尾部附睾的活精子数量及活力显著下降。然而给药PF562271 (30) 的小鼠对生育系统却没有显著影响。该研究表明, FAK的激酶依赖活性及支架功能分别具有独立的功能, 且支架功能在雄性动物的生殖系统中起到重要作用。未来是否可以基于PROTAC技术开发起效快, 生育能力可逆的高效男性避孕药还有待进一步研究。
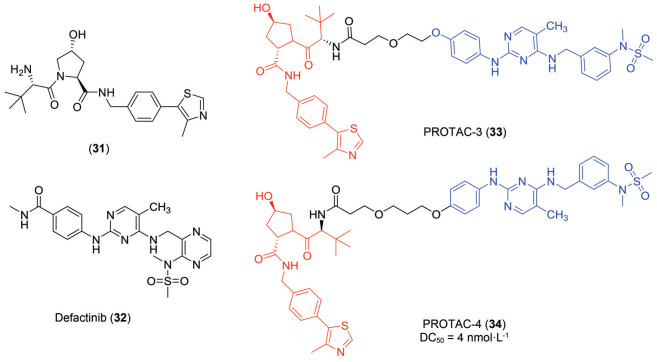
4.2 基于VHL靶向FAK的小分子PROTACsVon Hippel-Lindau (VHL) 是一种肿瘤抑制基因, 主要编码E3泛素连接酶pVHL, 该蛋白与Elongin B & C形成Cullin环E3泛素连接酶复合体, 再通过招募E2泛素结合酶, 识别蛋白质底物, 并使泛素转移到蛋白质底物上, 最终通过蛋白酶体系统被降解。VHL是首个广泛应用于研发小分子PROTAC的E3泛素连接酶配体[38]。常用的配体有化合物(31)。
2018年耶鲁大学Crews团队[39]为了研究FAK非激酶依赖的支架功能, 设计并合成了首个基于VHL配体结构新颖的FAK小分子PROTAC。该团队以defactinib (32) 为先导化合物, 通过构效关系研究(SAR) 发现, defactinib (32) 结构上的N-甲基苯甲酰胺暴露于蛋白口袋的亲水侧; 为了减少设计分子中的酰胺键, 增加细胞通透性, 作者用4-氨基苯酚来取代N-甲基苯甲酰胺; 为了降低合成难度, 作者用1, 3-二取代苯基来替代2, 3-二取代吡嗪基。因此, 研究人员在亲水侧引入不同长度的聚乙二醇(PEG) 连接链将"弹头"—defactinib衍生物与VHL配体连接, 最终合成出6个连接链长度各不相同的小分子FAK降解剂。正如预期的一样, 连接链和VHL配体的引入并不影响所设计的PROTAC对FAK激酶抑制活性的影响, 半数抑制活性(IC50) 均介于4.7~14.5 nmol·L-1。有趣的是抑制活性和降解活性没有相关性, 例如相比其他降解剂, PROTAC-4 (34) 的激酶抑制活性较差(IC50 = 14.5 nmol·L-1) 但降解活性却较好, 半数降解活性(DC50) 为4.0 nmol·L-1。对比其活性最强的PROTAC 3 (33) 与FAK抑制剂发现, PROTAC 3 (33) 的选择性要远大于defactinib (32)。在细胞迁移和入侵实验中表明, PROTAC 3 (33) 对MDA-MB-231细胞愈合抑制作用和抑制细胞迁移能力要强于defactinib (32)。该研究证明了PROTAC在扩大成药空间、增加选择性和控制蛋白质功能方面具有强大的潜力, 而这些功能是传统的小分子抑制剂难以具备的。
|
|
当前基于PROTAC技术用于靶向FAK的降解是一个全新的领域, 在2018年首次有可降解FAK的PROTAC的合成, 总的来说靶向降解FAK的报道并不是很多, 且作用机制的研究也不够深入。尽管人类基因组预测有600多种E3泛素连接酶[40], 但目前仅有几种被用于PROTAC的构建, 用于降解FAK的PROTAC的E3连接酶配体, 目前相关文献仅报道了VHL和CRBN, 但PROTAC技术正在快速发展, 相信不久会有更多的E3泛素连接酶配体被开发。
5 总结和展望FAK在癌症的发生、发展中起着关键作用, 被认为是抗癌药研发的重要靶点。尽管基于不同结构域的小分子抑制剂被大量研发, 且在临床上取得不错的治疗效果, 但是小分子抑制剂难以靶向FAK的支架功能只能抑制其激酶依赖活性, 这可能是限制FAK小分子抑制剂进一步临床使用的重要因素之一。为验证FAK靶点的成药性、研究其支架功能在生物体能的作用及更加深入地了解FAK靶点, 迫切需要一种新技术来快速、可逆的选择性降解FAK蛋白。PROTAC技术是近年来化学敲除致病蛋白的新型策略, 能同时阻断FAK的激酶依赖性功能和非激酶依赖的支架功能, 能解决小分子抑制剂只作用于蛋白激酶结构域带来的耐药性问题, 且和小分子抑制剂相比具有给药剂量低、毒副作用小的特点。PROTAC的独特作用机制及显著优势大大吸引了来自制药工业界及学术界的兴趣。特别是近年来具有口服活性的PROTAC进入临床研究且取得令人兴奋的试验结果, 为PROTAC的研究打开了新纪元。
尽管当前已有多个基于CRBN和VHL的靶向降解小分子PROTAC的报道, 且能选择性地降解FAK, 但目前的研究尚未表明PROTAC的抗癌活性优于FAK抑制剂, 且PROTAC在给药时遇到一系列问题, 比如脱靶、低细胞渗透性、不稳定性、不易吸收、不易合成和超大的分子量的诸多挑战[13, 41]。此外, E3泛素连接酶的选择对于FAK的降解至关重要, 当前仅有CRBN和VHL的配体成功应用于降解FAK PROTAC的设计, 未来还需要开发更多的E3泛素连接酶配体来实现PROTAC的理性药物设计。相信随着PROTAC的研究不断深入, 会研发出越来越多降解活性和抗肿瘤活性更好的FAK降解剂, 为深入研究FAK靶点提供有效的探针分子, 为靶向FAK的药物研发奠定理论基础。
作者贡献: 徐颖若负责综述的资料收集和文章撰写; 张沁松、鲍润菲和吴菁艺参与了部分内容的撰写、文献整理、图片制作及文章修改; 曾申昕负责文章的选题、思路和框架的提出以及文章修改和检查。
利益冲突: 本文无利益冲突。
| [1] |
Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications[J]. Nat Rev Cancer, 2014, 14: 598-610. DOI:10.1038/nrc3792 |
| [2] |
Demircioglu F, Wang J, Candido J, et al. Cancer associated fibroblast FAK regulates malignant cell metabolism[J]. Nat Commun, 2020, 11: 1290. DOI:10.1038/s41467-020-15104-3 |
| [3] |
Lv PC, Jiang AQ, Zhang WM, et al. FAK inhibitors in cancer, a patent review[J]. Expert Opin Ther Pat, 2018, 28: 139-145. DOI:10.1080/13543776.2018.1414183 |
| [4] |
Chauhan A, Khan T. Focal adhesion kinase-an emerging viable target in cancer and development of focal adhesion kinase inhibitors[J]. Chem Biol Drug Design, 2020, 97: 774-794. |
| [5] |
Cance WG, Kurenova E, Marlowe T, et al. Disrupting the scaffold to improve focal adhesion kinase-targeted cancer therapeutics[J]. Sci Signal, 2013, 6: pe10. DOI:10.1126/scisignal.6306er10 |
| [6] |
Ding Y, Fei Y, Lu B. Emerging new concepts of degrader technologies[J]. Trends Pharmacol Sci, 2020, 41: 464-474. DOI:10.1016/j.tips.2020.04.005 |
| [7] |
Wang Y, Jiang X, Feng F, et al. Degradation of proteins by PROTACs and other strategies[J]. Acta Pharm Sin B, 2020, 10: 207-238. DOI:10.1016/j.apsb.2019.08.001 |
| [8] |
Sun X, Gao H, Yang Y, et al. PROTACs: great opportunities for academia and industry[J]. Signal Transduct Target Ther, 2019, 4: 64. DOI:10.1038/s41392-019-0101-6 |
| [9] |
Luh LM, Scheib U, Juenemann K, et al. Prey for the proteasome: targeted protein degradation-a medicinal chemist's perspective[J]. Angew Chem Int Ed Engl, 2020, 59: 15448-15466. DOI:10.1002/anie.202004310 |
| [10] |
Ward RA, Fawell S, Floc'h N, et al. Challenges and opportunities in cancer drug resistance[J]. Chem Rev, 2021, 121: 3297-3351. DOI:10.1021/acs.chemrev.0c00383 |
| [11] |
Donovan KA, Ferguson FM, Bushman JW, et al. Mapping the degradable kinome provides a resource for expedited degrader development[J]. Cell, 2020, 183: 1714-1731.e10. DOI:10.1016/j.cell.2020.10.038 |
| [12] |
Deshaies RJ. Multispecific drugs herald a new era of biopharmaceutical innovation[J]. Nature, 2020, 580: 329-338. DOI:10.1038/s41586-020-2168-1 |
| [13] |
Zeng S, Huang W, Zheng X, et al. Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: recent progress and future challenges[J]. Eur J Med Chem, 2021, 210: 112981. DOI:10.1016/j.ejmech.2020.112981 |
| [14] |
Wang Y, Long J, Chang Q, et al. The application of small molecule PROTAC in researches of different targets[J]. Acta Pharm Sin (药学学报), 2020, 55: 446-452. |
| [15] |
Schaller MD, Otey CA, Hildebrand JD, et al. Focal adhesion kinase and paxillin bind to peptides mimicking beta integrin cytoplasmic domains[J]. J Cell Biol, 1995, 130: 1181-1187. DOI:10.1083/jcb.130.5.1181 |
| [16] |
Zhou J, Yi Q, Tang L. The roles of nuclear focal adhesion kinase (FAK) on cancer: a focused review[J]. J Exp Clin Cancer Res, 2019, 38: 250. DOI:10.1186/s13046-019-1265-1 |
| [17] |
Antoine M, Emilie S, Philippe C, et al. Targeting focal adhesion kinase using inhibitors of protein-protein interactions[J]. Cancers (Basel), 2018, 10: 278. DOI:10.3390/cancers10090278 |
| [18] |
Martínez T, Navajas L, Lietha D. FAK structure and regulation by membrane interactions and force in focal adhesions[J]. Biomolecules, 2020, 10: 179. DOI:10.3390/biom10020179 |
| [19] |
Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility[J]. Nat Rev Mol Cell Biol, 2005, 6: 56-68. DOI:10.1038/nrm1549 |
| [20] |
Frame MC, Patel H, Serrels B, et al. The FERM domain: organizing the structure and function of FAK[J]. Nat Rev Mol Cell Biol, 2010, 11: 802-814. DOI:10.1038/nrm2996 |
| [21] |
Yoon H, Dehart JP, Murphy JM, et al. Understanding the roles of FAK in cancer: inhibitors, genetic models, and new insights[J]. J Histochem Cytochem, 2015, 63: 114-128. DOI:10.1369/0022155414561498 |
| [22] |
Lee BY, Timpson P, Horvath LG, et al. FAK signaling in human cancer as a target for therapeutics[J]. Pharmacol Ther, 2015, 146: 132-149. DOI:10.1016/j.pharmthera.2014.10.001 |
| [23] |
Takahashi R, Sonoda Y, Ichikawa D, et al. Focal adhesion kinase determines the fate of death or survival of cells in response to TNFalpha in the presence of actinomycin D[J]. Biochim Biophys Acta, 2007, 1770: 518-526. DOI:10.1016/j.bbagen.2006.11.011 |
| [24] |
Cance WG, Golubovskaya VM. Focal adhesion kinase versus p53: apoptosis or survival?[J]. Sci Signal, 2008, 1: pe22. |
| [25] |
Cox BD, Natarajan M, Stettner MR, et al. New concepts regarding focal adhesion kinase promotion of cell migration and proliferation[J]. J Cell Biochem, 2006, 99: 35-52. DOI:10.1002/jcb.20956 |
| [26] |
Golubovskaya VM, Ho B, Zheng M, et al. Mitoxantrone targets the ATP-binding site of FAK, binds the FAK kinase domain and decreases FAK, Pyk-2, c-Src, and IGF-1R in vitro kinase activities[J]. Anticancer Agents Med Chem, 2013, 13: 546-554. DOI:10.2174/1871520611313040003 |
| [27] |
Thamilselvan V, Craig DH, Basson MD. FAK association with multiple signal proteins mediates pressure-induced colon cancer cell adhesion via a Src-dependent PI3K/Akt pathway[J]. FASEB J, 2007, 21: 1730-1741. DOI:10.1096/fj.06-6545com |
| [28] |
Liang C, Zhang N, Tan Q, et al. CT-707 overcomes resistance of crizotinib through activating PDPK1-AKT1 pathway by targeting FAK[J]. Curr Cancer Drug Targets, 2019, 19: 655-665. DOI:10.2174/1568009618666181031152140 |
| [29] |
Hood JD, Frausto R, Kiosses WB, et al. Differential alphav integrin-mediated Ras-ERK signaling during two pathways of angiogenesis[J]. J Cell Biol, 2003, 162: 933-943. DOI:10.1083/jcb.200304105 |
| [30] |
Lu Y, Sun H. Progress in the development of small molecular inhibitors of focal adhesion kinase (FAK)[J]. J Med Chem, 2020, 63: 14382-14403. DOI:10.1021/acs.jmedchem.0c01248 |
| [31] |
Moon S, Lee BH. Chemically induced cellular proteolysis: an emerging therapeutic strategy for undruggable targets[J]. Mol Cells, 2018, 41: 933-942. |
| [32] |
Edmondson SD, Yang B, Fallan C. Proteolysis targeting chimeras (PROTACs) in 'beyond rule-of-five' chemical space: recent progress and future challenges[J]. Bioorg Med Chem Lett, 2019, 29: 1555-1564. DOI:10.1016/j.bmcl.2019.04.030 |
| [33] |
Powell CE, Gao Y, Tan L, et al. Chemically induced degradation of anaplastic lymphoma kinase (ALK)[J]. J Med Chem, 2018, 61: 4249-4255. DOI:10.1021/acs.jmedchem.7b01655 |
| [34] |
Huang HT, Dobrovolsky D, Paulk J, et al. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader[J]. Cell Chem Biol, 2018, 25: 88-99e86. DOI:10.1016/j.chembiol.2017.10.005 |
| [35] |
Popow J, Arnhof H, Bader G, et al. Highly selective PTK2 proteolysis targeting chimeras to probe focal adhesion kinase scaffolding functions[J]. J Med Chem, 2019, 62: 2508-2520. DOI:10.1021/acs.jmedchem.8b01826 |
| [36] |
Gao H, Wu Y, Sun Y, et al. Design, synthesis, and evaluation of highly potent FAK-targeting PROTACs[J]. ACS Med Chem Lett, 2020, 11: 1855-1862. DOI:10.1021/acsmedchemlett.9b00372 |
| [37] |
Gao H, Zheng C, Du J, et al. FAK-targeting PROTAC as a chemical tool for the investigation of non-enzymatic FAK function in mice[J]. Protein Cell, 2020, 11: 534-539. DOI:10.1007/s13238-020-00732-8 |
| [38] |
Pettersson M, Crews CM. Proteolysis targeting chimeras (PROTACs) - past, present and future[J]. Drug Discov Today, 2019, 31: 15-27. DOI:10.1016/j.ddtec.2019.01.002 |
| [39] |
Cromm PM, Samarasinghe KTG, Hines J, et al. Addressing kinase-independent functions of FAK via PROTAC-mediated degradation[J]. J Am Chem Soc, 2018, 140: 17019-17026. DOI:10.1021/jacs.8b08008 |
| [40] |
Konstantinidou M, Li J, Zhang B, et al. PROTACs- a game-changing technology[J]. Expert Opin Drug Discov, 2019, 14: 1255-1268. DOI:10.1080/17460441.2019.1659242 |
| [41] |
Zeng SX, Huang WH, Shen ZR. Opportunities and challenges of PROTAC in R & D of small-molecule drugs[J]. Prog Pharm Sci (药学进展), 2020, 44: 801-816. |