 2021, Vol. 56
2021, Vol. 56

肌萎缩侧索硬化症(ALS) 属于罕见病, 为一种影响上运动神经元(大脑、脑干、脊髓) 和下运动神经元(颅神经核、脊髓前角细胞) 及其支配的躯干、四肢和头面部肌肉的一种慢性退行性疾病。临床上常表现为上下肢无力、消瘦并导致瘫痪, 著名物理学家霍金就是ALS患者。
ALS的病因有多种学说: 包括病毒感染侵入中枢, 铅、铝、铜等重金属中毒, 自身免疫, 遗传因素和兴奋性氨基酸等学说, 由于病因不清楚, 缺少有效的防治措施。研究表明, 氧化应激是ALS发作和进展的大概率因素, 氧化应激与脂质过氧化密切相关。在几乎没有药物可以延缓ALS恶化的情况下, 研发自由基捕获剂可能减轻氧化应激效应, 延缓ALS患者的神经功能的退化。美国FDA 2017年批准依达拉奉治疗ALS, 是FDA 22年来批准的首款ALS治疗药物。
1.2 抗脂质过氧化的自由基捕获剂其实依达拉奉的研制始自于治疗脑卒中患者。脑卒中导致局部脑缺血, 血液再灌注会启动酶促或非酶性产生活性氧(ROS), 活性氧多为羟基自由基、过氧化氢和其他过氧自由基。酶源性ROS的生成主要来自花生四烯酸发生的级联反应, 多以自由基形式存在。自由基的链反应产生过量的ROS, 引起能量衰竭和氧化应激, 使脑神经细胞膜损伤, 导致脑水肿和细胞死亡。缺血后的ROS炎性氧化应激还损伤血脑屏障(BBB), 引起血管性水肿加剧, 使水肿与细胞死亡进一步加剧(Banno M, Mizuno T, Kato H, et al. The radical scavenger edaravone prevents oxidative neurotoxicity induced by peroxynitrite and activated microglia. Neuropharmacology, 2005, 48: 283-290)。
酚类化合物是最早发现的自由基捕获剂, 实验表明, 酚类可以加速PGG2转变为PGH2, 从而使花生四烯酸的级联反应上调游离的PGI2 (Egan RW, Paxton J, Kuehl FA Jr. Mechanism for irreversible self-deactivation of prostaglandin synthetase. J Biol Chem, 1976, 251: 7329-7935), 例如化合物1可以阻止或减轻PGG1、PGE1等过氧化物对PGI2合成酶的抑制作用(Ham EA, Egan RW, Soderman DD, et al. Peroxidase-dependent deactivation of prostacyclin synthetase. J Biol Chem, 1979, 254: 2191-2194), 因而抑制炎症的发生和发展。
1.3 烯醇与酮的互变异构在化学上酚类具有捕获自由基的性质, 但作为药用, 由于多有不良反应和成药性的缺陷例如酚类对机体有腐蚀性和刺激性, 容易被代谢氧化和经II相代谢而失活, 因而难以直接应用。
|
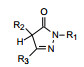
日本三菱制药的渡边等从化学观点分析, 以如下的论点设计自由基新型捕获剂: 酚类可视作烯醇, 经质子转移可互变成为酮式异构体, 从而引申一些含有α氢的环酮、内酰胺或环酰肼等可以烯醇形式捕获自由基的化合物, 起到酚类抗氧化的作用, 从而避免了酚类的毒性和易代谢性。图 1示意了该设计的思路。

|
Figure 1 Design of carbonyl compounds based on the phenol tautomerism (X=CH, N, etc) |
用离体的硫代巴比妥酸(TBA) 法测定受试化合物对大鼠脑匀浆自氧化的抑制实验, 评价化合物活性。不同浓度的受试化合物与大鼠脑匀浆温孵30 min, 离心后的上清液加入十二烷基硫酸钠(SDS, 应称作硫酸十二醇酯钠), 酸化, 加入TBA, 加热, 萃取, 荧光测定TBA的强度, 计算抑制50%自氧化反应的受试物浓度(IC50)。
体内评价抗脑缺血活性用大鼠造模。实验组腹腔注射10 mg·kg-1化合物, 对照组注射生理盐水, 30 min后将左颈总动脉、左锁骨下动脉和胸腔的头臂动脉同时钳夹, 致使脑电波(EEG) 立即变平, 钳夹10 min, 使恢复循环, 记录EEG恢复时间和动物存活时间。
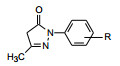
3 化合物结构及优化 3.1 吡唑烷酮的N1-取代基变换目标化合物需要穿越BBB进入中枢, 淬灭脑缺血引起的脂质过氧化所产生的自由基, 分子尺寸宜小和适宜的亲脂性, 以有利于穿越BBB向脑中分布。研发者设计了吡唑烷酮为骨架的分子, 4位至少含有一个氢原子, 以便形成烯醇型分子。首先合成了容易制备的化合物2 (乙酰乙酸酯与苯肼缩合而得), 缺血大鼠模型实验表明, 2可恢复消失(变平) 的脑电波, 提示具有脑保护作用(Watanabe T, Morita I, Nishi H, et al. Preventive effect of MCI-186 on 15-HPETE induced vascular endothelial cell injury in vitro. Prostaglandins Leukot Essent. Fatty Acids, 1988, 33: 81-87)。从而以化合物2为先导物, 首先变换N1取代基, 合成的化合物及其活性列于表 1。
| Table 1 The effect of alteration of N1-substituents on the lipid peroxydation. * Inhibitory percentage at 500 μmol·L-1, and the same below |
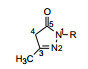
表 1的构效关系表明, N1的苯基用氢或甲基(3和4) 等小位阻基团置换, 丧失了活性, 羟乙基(5) 也无活性, 可能是亲脂性低的缘故, 推论抗脂质过氧化需有一定的亲脂性。环己基(6)、苄基(7)、1-萘基(8) 或杂环(9~13, 10除外) 都保持或提高了活性, 1-萘基和2-苯并噻吩基化合物的体外活性高于2, 可能是亲脂性较强所致。由于2仍是活性较好的化合物, 下一步是在苯环上进行取代。
3.2 N1-苯环上的取代基变换在苯环的不同位置作各种基团取代, 化合物及活性列于表 2。分析构效关系表明: ①苯环的2位取代大都使活性显著减弱, 如化合物14、21和30, 2-OH取代(25) 活性尚可, 可能是酚羟基贡献于抗氧化作用, 也可能是与N2形成分子内氢键, 消除了邻位取代的位阻效应。②苯环的3位与4位取代基对活性没有显著差异。③增加基团的亲脂性如烷基和卤素可提高活性, 长链的烷基或烷氧基也使活性增高。④二氯化合物(24) 活性强于单取代物, 而二甲氧基的活性并未提高。⑤极性基团如羟甲基(29) 和羧基(35) 活性降低或丧失。综上, 在N1的苯环3或(和) 4位含有亲脂性基团有利于增强抗脂质过氧化活性。
| Table 2 The structure and the anti-lipid peroxidation activity of the compounds with varied substituent on the phenyl ring |
吡唑环的N2和5-酮基是不可变换的。下一步是变换吡唑环的3位和4位基团, 合成的化合物活性列于表 3。构效关系显示: ①增加3位基团的亲脂性, 化合物44和45的抗脂质过氧化活性增高, 含有吡啶环的化合物47和49具有中等活性, 而苯基取代的46活性异常高, 强于有极性的呋喃化合物48大约50倍, 提示3位需有亲脂性基团。② 4位的亲脂性也有利于抗氧化活性。5-异丁基38提高了活性, 而5-羟乙基39几乎没有活性。苯氧基(40) 和苯硫基(41) 虽然亲脂性较强, 但活性未能提高(或许是位阻大的原因)。化合物42是4, 4-二甲基化合物, 丧失了活性, 是由于没有α氢, 不能形成烯醇的缘故, 证实了酮-烯醇互变异构是这类化合物捕获自由基的关键因素。
| Table 3 3- and 4-Substituted pyrazolidones and the anti-lipid peroxidation activity |
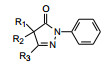
表 1~3中多数化合物体外实验具有抑制脂质过氧化活性, 但体内大鼠脑缺血模型表明只有9个化合物呈现活性, 表 4列出了这些化合物的体内活性, 包括脑电图恢复的时间(时间越短活性越强) 和大鼠存活的时间(时间越长活性越强)。结果表明, 空白对照组动物未能恢复脑电图, 大鼠存活的时间最短, 受试化合物的存活时间是不同的。其中化合物2、32和34表现为优良的化合物。
| Table 4 In vivo activity of the active compounds |
对高活性化合物的进行成药性实验, 表明化合物2优胜于其他分子。静脉注射2, 在血浆中50%以烯醇的阴离子形式存在, 这是捕获自由基的活性形式。60%分布到中枢, 血浆半衰期4.5~6 h, 代谢物为Ⅱ相代谢硫酸氢酯和葡醛酸苷, 没有活性。2定名为依达拉奉(edaravone)。三菱制药将依达拉奉进入日本临床试验, 治疗因卒中引起的脑水肿, 于2001年批准在日本上市。自2011年开始对ALS的Ⅲ期临床试验, 日本于2015年批准为缓解ALS新的适应症。2017年5月美国FDA根据日本治疗ALS的效果, 作为孤儿药直接批准在美国上市, 治疗肌萎缩侧索硬化症(Watanabe K, Morinaka Y, Iseki K, et al. Structure-activity relationship of 3-methyl-1-phenyl-2-pyrazolin-5-one (edaravone). Redox Reports, 2003, 8: 151-155; Watanabe T, Tahara M, Todo S. The novel antioxidant edaravone: from bench to bedside. Cardiovasc Ther, 2008, 26: 101-114)。
|
依达拉奉的pKa = 7.0, 在生理条件下, 经互变异构50%可成为烯醇式阴离子, 与体内自由基(例如脂质过氧化生成的氧自由基) 经单电子转移, 生成依达拉奉自由基, 后者与分子氧反应, 经依达拉奉过氧自由基生成二酮化合物, 水解得到终产物丁二酮酸苯腙。二酮还可逆地与另一分子依达拉奉缩合生成羟基二聚物。图 2是依达拉奉清除自由基的反应机制(Yamamoto T, Yuki S, Watanabe T, et al. Delayed neuronal death prevented by inhibition of increased hydroxyl radical formation in a transient cerebral ischemia. Brain Res, 1997, 762: 240-242)。

|
Figure 2 The mechanism of free-radical scavenge of edaravone |
依达拉奉抑制人嗜中性白细胞产生的活性氧(例如过氧化氢和羟基自由基等) 是通过上述的反应机制实现的, 而不是抑制细胞的功能所致(Mikawa K, Akamatsu H, Nishina K, et al. Effects of edaravone on human neutrophil function. Acta Anaesthesiol Scand, 2005, 49: 385-389)。
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
肌萎缩侧索硬化症(ALS) 是难治的罕见病, 没有有效的治疗药物。美国FDA根据日本临床研究的数据直接批准上市, 是20多年来批准的首款ALS新药。三菱制药研制依达拉奉最初为治疗和缓解脑卒中后遗症, 是从消除脑内活性氧自由基入手的, 以化学视角淬灭自由基设计化合物。从酚类抗氧化作用提出了烯醇-酮的互变异构的科学假设, 设计出吡唑啉酮骨架, 经过体内外构效关系的实验求证, 优化出超小分子(MW = 174) 药物依达拉奉, 具有强效捕获自由基的物化性质和适宜的亲脂性, 因而易于穿越BBB和有利于清除脑内自由基。这个研发路径值得借鉴。 (编者按)
(·新药发现与研究实例简析·)