 2021, Vol. 56
2021, Vol. 56

间变性淋巴瘤激酶(ALK) 是受体酪氨酸激酶中胰岛素受体家族的成员, 在正常状态下主要存在于神经组织中, 生理功能尚不清楚, 可能与中枢和外周神经系统发育有关。然而某些全身性肿瘤所发生的t(2;5) (p23;q25) 染色体易位, 产生了ALK与核磷蛋白(nucleophosmin, NPM) 胞内结构域的融合基因NPM-ALK, 高表达的NPM-ALK蛋白是发生间变性大细胞淋巴瘤(ALCL)、非小细胞肺癌(NSCLC) 等关键性酶, 因而是研发这类疾病治疗药物的重要靶标。
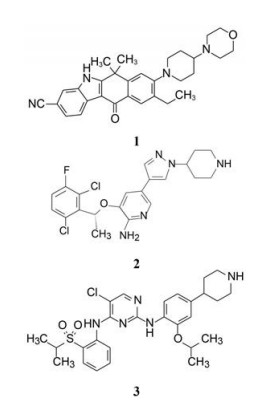
1.2 ALK抑制剂的研发艾乐替尼(1, alectinib) 是由日本中外药业原研, 罗氏公司开发, 于2014年7月经美国FDA批准上市的ALK抑制剂。在此之前辉瑞研发的克唑替尼(2, crizotinib) 于2011年上市, 是第一个ALK激酶抑制剂。诺华研发的色瑞替尼(3, ceritinib) 于2014年4月上市。艾乐替尼和色瑞替尼认为是作用于ALK的第二代药物。虽然是后继性产品, 鉴于新药研发的长周期性, 都具有独立研发的特征。
2 生物学评价 2.1 酶抑制活性评价受试化合物的抑制ALK酶活性是在30 μmol·L-1浓度的ATP和10 mmol·L-1的MgCl2存在下, 用时间分辨荧光共振能量转移测定法(TR-FRET) 测定不同浓度的受试物抑制ALK对底物蛋白(生物素-EGPWLEEEEEAYGWMDF) 的磷酸化程度, 计算半数抑制浓度IC50。
|
用具有高表达NPM- ALK蛋白、ALK呈阳性的人肿瘤Karpas 299细胞, 与不同浓度的受试物温孵96 h, 用细胞活力检测试剂盒(CCK-8) 测定对细胞生长的抑制作用, 计算半数抑制浓度IC50。
2.3 对肝微粒体的稳定性实验含有浓度为1 mmol·L-1 NADPH的50 μmol·L-1磷酸缓冲液中, 加入1 μmol·L-1受试物和人(或小鼠) 肝微粒体(0.5 mg·mL-1), 于37 ℃温孵30 min, 加入乙腈终止反应, 离心, 上清液用LC/MS定量测定受试物的含量, 评价化合物对微粒体的稳定性(CL), 单位是μL·min-1·mg-1。
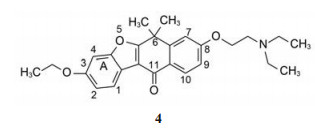
3 先导化合物及其优化 3.1 先导物的发现为发现抑制ALK的先导化合物, 中外公司普筛了激酶抑制剂的化合物库, 发现了四环化合物4具有初步活性, IC50为1.3 μmol·L-1。并通过Linweawer-Burk双倒数作图表明对ATP竞争性结合的Ki = 0.17 μmol·L-1, 化合物4对c-Met激酶的活性IC50 > 50 μmol·L-1, 提示对ALK有较高的选择性作用, 这是该项目所设定的目标。
|
换变换3-位取代基以考察烷氧基对酶活性的影响, 不改变其他位置的取代基如6, 6-二甲基、11-酮基和8位的二乙胺乙氧基, 以免增加构效分析的复杂性。表 1列出的数据表明, 3-烷氧基的碳数对活性没有显著影响(4~6)。而烷氧基被卤素取代活性明显升高(7~9), 尤其是氯原子取代的化合物(9) 活性比先导物4强2倍。但拉电子的三氟甲基的活性反而减弱, 提示基团的电性作用不是影响活性的因素。
| Table 1 3-Substituted tetracyclic compounds and their activities against ALK |

将四环骨架中的苯并呋喃换成吲哚, 考察呋喃环中的氧原子(氢键接受体) 变换成NH (氢键给体) 对活性的影响, 同时对3位取代基进行优化, 不改变其他位置的基团。结构与活性列于表 2。
| Table 2 Activities of compounds containing indole fragments in the framework |
表 2的结果表明, 苯并呋喃换作吲哚的化合物活性显著提高, 例如11的活性强于8大约10倍, 3-氯取代物(13) 也强于相应的9。吲哚的氮推电子性强于氧原子, 可能提高了11位酮基与酶铰链区形成氢键的能力。3-氰基化合物(14) 的活性尤其高, IC50达到纳摩尔水平, 可能是CN形成氢键, 提高了结合能力(烷氧基的氧原子位置与CN的氮不同)。这些活性较高的化合物对高表达NPM-ALK的Karpas 299细胞也具有抑制活性, 而且与酶活性呈相关性。
3.4 8位侧链的变换化合物14对小鼠和人肝微粒体代谢迅速, 是由于侧链的二乙胺基发生氧化去烷基作用。为了优化代谢稳定性, 将3-氰基吲哚片段固定不变, 改变8位侧链的结构, 代表性的化合物列于表 3。
| 表 3 Activities of compounds with changed 8-position side chain |








分析表 3化合物的构效关系如下: ①用磺酰基环合的化合物(15) 仍保持较高的活性, 但未能提高代谢的稳定性。②侧链为酰胺(16) 和脲(17) 的稳定性显著提高, 但抑制细胞增殖作用减弱。③经氮原子连接的环状片段(18~20), 抑制酶和细胞活性都减弱, 氮上的电子离域化到苯环, 侧链上没有碱性氮原子, 提示碱性氮对结合于ALK酶的必要性, 因而是呈现活性的重要因素。④ 3个哌嗪化合物21~23中21没有可形成正离子的氮原子, 抑制细胞活性弱, 而22和23活性强。而且含有氧杂环丁基的化合物23有较低的肝清除率。其实, 氧杂环丁烷片段因具有良好的物化性质, 在药物设计颇受重视(Wuitschik G, Carreira EM, Wagner B, et al. Oxetanes in drug discovery: structural andsynthetic insights. J Med Chem, 2010, 53: 3227-3246)。⑤化合物23的哌嗪环用哌啶取代得到化合物24, 24的抑制酶和细胞活性强于23, 并维持代谢稳定性, 这对以后的优化提示了线索。
3.5 9位取代基的优化化合物23有优良活性和稳定性的品质, 进而评价了对17种激酶的活性, 表明对多数激酶的抑制活性很弱, 提示对ALK有较好的选择性。然而对KDR等激酶仍显示活性, 为消除脱靶作用的风险继续作结构优化(Kinoshita K, Ono Y, Emura T, et al. Discovery of novel tetracyclic compounds as a anaplastic lymphoma kinase inhibitors. Bioorg Med Chem Lett, 2011, 21: 3788-3793)。
23的抑制活性是由于竞争性地结合ALK激酶的ATP位点, 由于人体已知的470个激酶中, 只有14个E0位点是由两个小尺寸氨基酸构成, 其中包括ALK, E0位点是Ala1200-Gly201 (Liao JJL. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J Med Chem, 2007, 50: 409-424), 这与其他相关酶的E0的组成不同。表 4列出了ALK和脱靶蛋白的E0和其他重要位点的组成。可以看出只有ALK的E0区体积较小。这为分子设计提高选择性提供了结构依据。
| Table 4 Composition of important sites and E0 of ALK and related kinases |
分子模拟显示, ALK的Ala1200-Gly201区域是配体的入口, 由于氨基酸的尺寸小留有较宽的结合面, 因而对应于23的9位可加入亲脂性基团以与Ala1200-Gly201结合, 而其他激酶的E0位点空间减小, 加入的基团形成位阻, 不利于结合, 从而可提高选择性抑制ALK, 减少脱靶作用。图 1为23与ALK的分子对接图, 显示9位与Ala相距4.2 Å, 有引入基团的空间。

|
Figure 1 Molecular docking diagram of compound 23 and ALK crystal structure |
设计合成的9-取代化合物的结构与抑制ALK和脱靶激酶(KDR、KIT和MET) 的数据列于表 5。构效关系表明, 9-甲基(25) 对ALK和脱靶蛋白的作用影响不大, 而增大烷基尺寸(26~30) 明显降低了与脱靶蛋白的结合, 说明大体积基团对脱靶蛋白的位阻作用。其中乙基(26) 和乙炔基(27) 是分辨活性与选择性的最适基团。这些化合物对人肝微粒体的耐受性除环戊乙基(30) 外都比较好。
| Table 5 Activities of compounds with 9-position substitution |
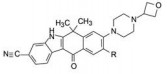
用17种激酶对化合物27的选择性和猴药代动力学做深入研究, 表明是优良的化合物, 并用移植KARPAS-299细胞的小鼠做体内抗肿瘤实验(灌胃2、6和20 mg·kg-1, 每日一次连续11天), 显示有剂量依赖性的强效抗肿瘤效果, 即使高剂量(20 mg·kg-1) 灌胃给药, 也未见明显的体重减轻, 提示不良反应较小(Kinoshita K, Kobayashi T, Asoh K, et al. 9-Substituted 6, 6-dimethyl-11-oxo-6, 11-dihydro-5H-benzo[b]carbazoles as highly selective and potent anaplastic lymphoma kinase inhibitors. J Med Chem, 2011, 54: 6286-6294)。
4 8位和9位的基团优化 4.1 9位含氮正离子的优化前已述及, 8位含有可形成正离子的侧链有利于对ALK的结合(含可质子化的哌嗪或哌啶环), 9位存在有一定体积的基团有利于ALK的选择性作用。研发者将优化出的碱性侧链连接于9位, 将提高活性强度和选择性两个因素集合在一个位置上, 合成代表性化合物列于表 6。
| Table 6 Activity, selectivity and metabolic stability of 9-substituted compound |

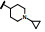
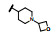
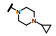

表中数据给出如下信息: 与脱靶蛋白相比, 9-含氮侧链对ALK保持有选择性, 然而对ALK的活性强度低于化合物27, 提示9位的环状片段的体积有碍于同ALK的结合(体积过大)。在对肝微粒体的稳定性上, 化合物31和33与相应的氧杂环丁烷片段没有显著差异, 揭示连接到8位的侧链可以不拘泥于氧杂环丁基。
4.2 9-乙基-8-取代基的再优化既然9位疏水性取代基不能过大, 再回到8, 9位双取代的模式, 固定9-乙基, 变换8位的含氮侧链, 合成的代表性化合物列于表 7。
| Table 7 Activities of 8-substituted 9-ethyl compounds |

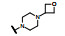


结果表明, 将制约活性强度的基团与决定选择性的基团分别连接于8位和9位, 活性强度与选择性都显著强于合并于9位的化合物, 而且对肝微粒体的稳定性也加强了, 说明9-乙基阻止了微粒体对8位的氧化代谢; 也提示氧杂环丁基可更换成其他脂环或杂环。化合物1为吗啉取代基, 对KARPAS-299细胞的抑制活性显著高于其他化合物, 出乎意料, 原因不详。1对肝微粒体的稳定性也很高。
4.3 9位烷基的再优化化合物1揭示了8-吗啉基哌啶的新片段, 是否9-乙基仍是最佳基团的配置, 需再审视(这是优化的常规操作), 为此固定8位不变, 考察9位烷基取代对活性、选择性和稳定性的影响, 合成的代表性化合物列于表 7。
结果表明化合物1是优胜的化合物, 对细胞抑制活性最强, 选择性最高, 对人和小鼠肝微粒体的稳定性最好, 37~39都在某种(些) 性质上逊于1, 从而确定1为候选化合物进入开发阶段(Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem, 2012, 20: 1271-1280)。
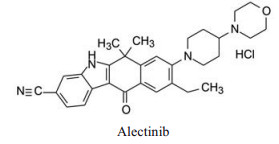
5 候选物艾乐替尼的上市化合物1进一步扩大对37种激酶的活性评价, 表明只对ALK激酶呈现高抑制活性, 其余为低或无活性, 表现出高度选择性作用。对NMP-ALK阳性的间变性大细胞淋巴瘤(ALCL) 细胞移植的小鼠模型和EML4-ALK阳性的非小细胞肺癌(NSCLC) 细胞移植的小鼠模型灌胃1 (20 mg·kg-1), 肿瘤生长抑制作用(TGI) 分别为119%和168%, 疗效显著。大鼠和猴的药代动力学实验表明口服生物利用度F分别为65.2%和50.4%, 半衰期t1/2分别为12.6和8.38 h, 静脉注射的清除率CL分别为7.79和6.04 μL·min-1·mg-1。遂以盐酸盐形式进入临床研究, 定名为艾乐替尼(alectinib), 经三期临床研究证明对晚期(转移性)A LK阳性非小细胞肺癌是有效口服治疗药, 于2014年经美国FDA批准上市。
|
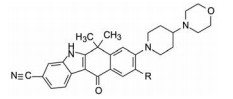
| Table 8 Activity of 9-position analogue of compound 1 |
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
艾乐替尼是以间变性淋巴瘤激酶为靶标治疗非小细胞肺癌的第三个上市药物, 与首创药物克唑替尼相距两年多, 和第二个色瑞替尼只相差三个月, 从迥异的分子结构可以判断他们都是独立研制成功的。这是个以靶标为核心和出发点的创新药物, 涵盖了通量普筛、结构优化, 优化包括对酶和细胞的活性强度、选择性作用和代谢稳定性等, 应用分子模拟和试错性(trail and error) 构效分析与反馈, 显示了传统药物化学和理性设计的结合。然后通过整体动物的药效药代和安全性评价, 过渡到临床的概念验证和安全有效性评价, 展现出完整的新药创制过程。(编者按)
(·新药发现与研究实例简析·)