 2021, Vol. 56
2021, Vol. 56

由于内在或外来的作用, 细胞内DNA处于不断地损伤和修复中。受损伤的单链DNA由聚ADP核糖聚合酶(PARP)催化修复, 双链的由BRCA修复, BRCA失效(例如突变)时PARP也履行修复功能。动物敲除PARP-1基因, 失去修复的功能, 经射线照射或细胞毒药物处理引起的细胞损伤会显著加重。许多化疗药物的作用机制是诱导DNA损伤, 而癌细胞又通过自身的PARP和BRCA修复因而产生耐药性。所以化疗后用PARP抑制剂治疗, 使癌细胞“雪上加霜”。
聚ADP核糖聚合酶是由18个蛋白组成的家族, 参与细胞的的多种功能, 包括DNA修复、基因表达、细胞周期控制、能量代谢和细胞内移行等。PARP-1和PARP-2蛋白主要功能是修复作用(正常细胞和癌细胞), 具有DNA结合域, 催化烟酰胺腺嘌呤二核苷酸(NAD+)发生核苷化, 对碱基切除修复(BER), 修复DNA。
BRCA-1和BRCA-2是癌抑制因子, 参与修复DNA双螺旋损伤的关键成分。BRCA-1和BRCA-2的缺失或突变是多数乳腺癌和卵巢癌患者的特征。BRCA突变对癌细胞是双刃剑, 一方面, 因为它能更快地积累基因突变, 进化的更快, 更容易产生耐药性, 这是优势。另一方面, 因为已经失去BRCA功能的癌细胞, 如果PARP再被抑制, 就彻底失去了修复DNA的能力, 癌细胞就会很快死亡, 这是癌细胞的劣势。本项目的研发目标是抑制PARP和对BRCA发生变异的肿瘤抑制剂。
2 活性评价 2.1 化合物体外对PARP-1活性体外评价化合物对PARP-1的抑制作用是用闪烁近邻分析方法测定。在96孔板上将系列浓度的受试物加入到PARP-1酶与含有[3H]-NAD+的NAD+培养液中, 温孵后加入DNA底物, 3 h后加入链霉亲和素-闪烁近邻分析微珠(streptavidinSPA bead)终止反应, 5 min后用微板闪烁计数仪测定放射性强度, 由不同浓度的抑制率计算受试物的IC50。
2.2 测定受试物抑制聚ADP核糖化作用96孔板上接种HeLa细胞, 温孵生长后, 加入系列浓度的受试物, 温孵18 h, 加入H2O2水溶液(阴性对照组不加H2O2), 5 min后, 除去介质, 细胞用-20℃甲醇固定20 min, 去除固定液, 洗涤后加入初级单抗、二次抗鼠抗体和核染料, 暗室中温孵后去除溶液, 洗涤细胞后, 用InCell1000仪读数。测定PARP阳性核与被全部染色的细胞核的比值, IC50是基于残留的酶活性计算而得。
2.3 对细胞的作用用含有抗BRCA-1的短发卡RNA (shRNA)表达元件和绿色荧光蛋白表达元件的慢病毒转染HeLa细胞, 产生80%以上BRCA-1沉默的HeLa细胞, 在96孔板与不同浓度的受试物温孵7天, 用荧光计测定细胞生长的百分率。对照实验是野生型HeLa细胞, 只转染绿色荧光蛋白表达的细胞。计算受试物抑制半数细胞生长的浓度(IC50)。
2.4 体内活性评价雌性CD1小鼠接种人乳腺癌细胞(ATCC), 肿瘤生长到150 mm3, 每日灌胃100 mg·kg-1一次, 或50 mg·kg-1灌胃两次, 连续33天, 期间每周测量体重和肿瘤尺寸。
3 先导化合物 3.1 辅酶NAD+的作用PARP-1催化修复过程, 需有辅酶烟酰胺腺嘌呤二核苷酸(NAD+)参与, 本项目研发的策略依据, 是抑制剂占据NAD+结合位置, 与NAD+发生竞争性结合, 阻断反应的进行。X射线晶体结构和分子模拟显示, NAD+结构中烟酰胺的酰胺基与PARP-1的Ser904羟基和Gly863骨架酰胺形成3对氢键, 吡啶环与Tyr907发生π-π叠合作用。基于这种结合模式, 苗头化合物的筛选就是从具有这种结合特征开始的。
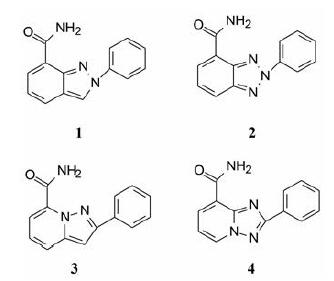
3.2 先导物的设计和确定先导物不是随机筛选而是设计得到的。为了满足上述的结合特征, 设计合成了[6,5]双杂环酰胺1~4, 杂环和酰胺基对应于NAD+的烟酰胺片段, 以形成氢键和π-π相互作用。活性评价显示具有中等活性(表 1), 以化合物1为最强, IC50=24 nmol·L-1, 而且也呈现细胞活性(EC50=3.7μmol·L-1), 提示可抑制因H2O2引起DNA损伤的PAR聚合物生成。由于1的较强活性确定为先导化合物。
|
| 表 1 Activity of the compounds optimized for initial skeleton |
化合物1作简单取代, 例如苄基或邻、间或对位取代苯基, 合成的化合物列于表 1。活性评价表明苄基(5) 或邻氯取代(6) 使活性降低, 而间(7)或对位(8) 氯代, 都提高了活性。邻位取代可能因位阻效应引起的构象变化不利于结合, 因而后继优化的位点在间或对位。此外, 化合物1的溶解性低(26 μg·mL-1), 7和8的溶解性更低, 不利于口服吸收, 后续的结构变换还须调整物化性质。
4 结构优化: 同时提高活性、选择性和物化性质在苯环对位连接出侧链和片段, 预计不仅不会影响母核与烟酰胺结合腔的结合, 而且连接出的脂肪胺(环胺或链胺)可能增加同腺苷位点的结合, 提高结合强度和抑制作用, 同时也因引入碱性基团提高分子的溶解性, 优化成药性。为此, 合成的化合物列于表 2。
| 表 2 Inhibitory activity of 2-substituted phenyl-2H-indazole-7-carboxamide on the proliferation of PARP and HeLa cells.a:Mutant BRCA;b:Wild BRCA |
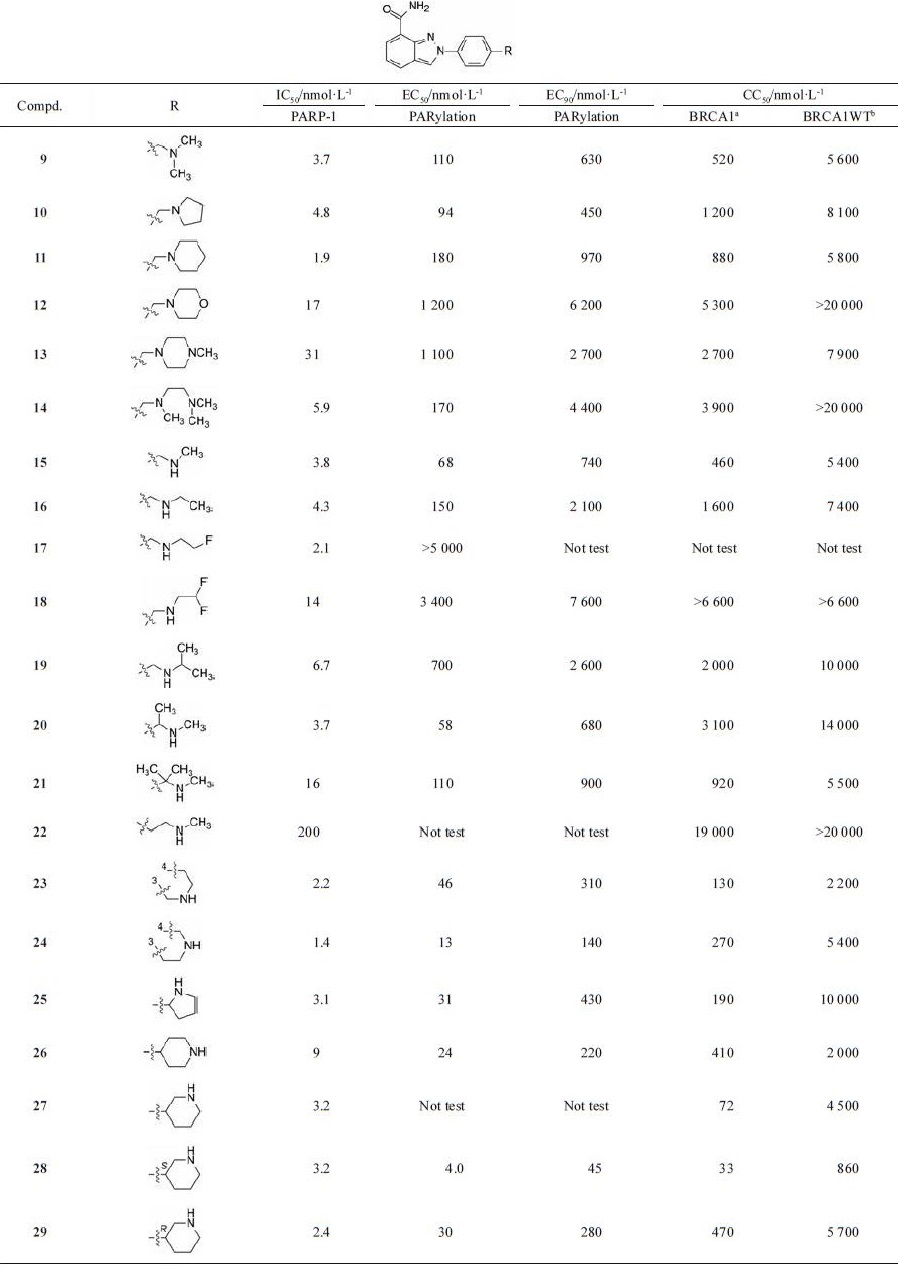
分析表 2的构效关系如下:①苯环对位连接二甲胺甲基, 化合物9的抑酶活性(IC50 = 3.7 nmol·L-1) 比先导物提高了6倍, 而且对DNA损伤修复生成聚ADP-核糖链的抑制活性提高了33倍。9对BRCA-1突变的子宫癌HeLa细胞抑制活性(CC50 = 520 nmol·L-1) 比没有缺失的细胞(CC50 = 5 600 nmol·L-1) 活性高10倍, 提示可选择性抑制BECA-1变异的细胞生长。②末端胺基成环, 化合物10和11仍保持活性, 对BRCA-1变异的细胞选择性为5倍于非突变株。吗啉环或甲基哌嗪环或开环的12~14活性稍弱。③仲胺15的抑酶活性与9相当, 抑制BRCA-1变异细胞的选择性为10倍。但N-乙基或异丙基的活性和选择性差。④活性较好的化合物15是经一个碳原子连接位阻小的仲胺, 但经两个碳相连的22活性显著下降。⑤合成的四氢吡咯、哌啶(分别与苯环以单键相连或并环) 等化合物, 是为了增加α碳的位阻, 降低CYP氧化代谢的不稳定性(下节叙述)。结果不仅增加了稳定性, 而且大多提高了活性和选择性。其中化合物27对PARP-1酶和BRCA-1突变的HeLa细胞活性与选择性显著提高, 进而将27拆分成光学异构单体, S构型(28) 优胜于R (29), S对变异细胞的活性强于R构型15倍, 提示哌啶环与靶酶的结合具有立体选择性。
5 评价药代动力学对活性较强的化合物评价了体内外药代性质, 列于表 3。其实, 诸轮设计合成的化合物不仅基于活性的变化, 还注意提高代谢稳定性。例如为了降低化合物15被大鼠肝微粒体清除率(CL = 28 μL·min-1·g-1), 避免苯基α碳被氧化, 在α位引入甲基或偕甲基, 化合物20和21基本保持活性, 由于增加了位阻, 提高了稳定性, 清除率CL分别为9和11 μL·min-1·g-1。四氢吡咯和哌啶经碳原子连接于苯环上, 也是为了提高稳定性。
| 表 3 In vivo and in vitro pharmacokinetic properties of active compounds. aRat liver microsomes; bHuman liver microsomes |
表 3中提示化合物20大鼠血浆清除率CL = 58 μL·min-1·kg-1, 表明稳定性高于没有α甲基的化合物15 (CL = 220 μL·min-1·kg-1)。
化合物27为3-哌啶化合物, 含有稳定的苯乙胺片段, 对大鼠肝微粒体和重组的CYP1A1的清除率分别为11和0.3 μL·min-1·g-1, 口服生物利用度F = 74%。S异构体(28) 大鼠体内的清除率CL = 28 μL·min-1·kg-1, 口服生物利用度F = 65%。R构型的CL = 24 μL·min-1·kg-1, 口服生物利用度F = 47%, S构型对BRCA-1突变的HeLa细胞活性CC50 = 33 nmol·L-1, 阴性对照的细胞毒作用CC50 = 860 nmol·L-1, 选择性为17倍; 而R构型对BRCA-1突变的HeLa细胞活性CC50 = 470 nmol·L-1, 阴性对照组CC50 = 5 700 nmol·L-1, 所以, S构型的28是药效与药代都优良的化合物。
6 候选化合物的确定和尼拉帕尼的上市进一步评价28对PARP亚型的抑制活性, 发现只选择性地对PARP-1和-2有强抑制活性(PARP-1 IC50 = 3.2 nmol·L-1; PARP-2 IC50 = 2.1 nmol·L-1), 对其他亚型PARP的IC50都大于330 nmol·L-1。抑制BECA-1发生变异乳腺腺癌细胞CC50 = 18 nmol·L-1; BECA-2发生变异的胰腺腺癌细胞活性为CC90 = 90 nmol·L-1, 而对未发生变异的上皮细胞无细胞毒作用。
化合物28的甲苯磺酸盐有良好的药代动力学性质, 大鼠口服生物利用度F = 65%, 较高的分布容积Vdss = 6.9 L·kg-1, 半衰期t1/2 = 3.4 h。
体内药效学实验表明, 雌性CD1裸鼠接种BRCA-1变异的MDA-MB-436细胞, 肿瘤生长到150 mm3, 每日灌胃100 mg·kg-1一次, 或50 mg·kg-1灌胃两次, 连续33天, 两种给药方法都使肿瘤消退, 体重减轻在10%以内, 裸鼠未出现死亡。

|
基于以上性质, 默克公司(后转给Tesaro公司) 确定化合物28为候选化合物, 定名为尼拉帕尼(niraparib), 经临床前和临床研究, 证明对BRCA1/2基因突变的卵巢癌和乳腺癌患者化疗后, 每日口服尼拉帕尼一次, “中位无进展生存时间”是21个月。而对照组使用安慰剂的病人, 只有5.5个月。由于疗效显著美国FDA于2017年3月批准上市。
7 后记尼拉帕尼是聚ADP核糖聚合酶(PARP) 抑制剂, 抑制肿瘤细胞PARP对受损伤DNA的修复, 成为带有BRCA基因突变的癌症的靶向治疗药物。国内外与尼拉帕尼同时研发PARP抑制剂的单位很多, 历程并不顺利, 例如第一个PARP抑制剂药物是阿斯利康2014年底上市的奥拉帕尼(olaparib), 几经周折成为卵巢癌治疗的四线用药。而尼拉帕尼的原研者默克公司于2008~2012年四年的临床研究未果而放弃, 2012年5月将其转让给Tesaro公司。Tesaro在卵巢癌和乳腺癌开展了精准性研究, 其关键性临床试验(NOVA) 是PARP抑制剂领域内Ⅲ期研究中最为成功的一个, 美国FDA也因此将尼拉帕尼列入快速通道进行优先审批。尼拉帕尼的成功使原本低迷的Tesaro股价暴涨, 体现了生物制药界的高风险高回报的常态。
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
尼拉帕尼是一个PARP基因的靶向药物, 主要针对BRCA1/2基因突变的卵巢癌和乳腺癌。从构建化学结构和活性评价乃至验证临床适应症等都紧紧瞄准发生BRCA1/2基因突变的PARP靶标, 彰显出精准医学的特点。先导物的设计基于参与PARP催化反应的辅酶I的部分结构, 优化过程中酶和细胞水平评价化合物的活性则针对突变的蛋白, 更重要的是Tesaro公司从默克接手项目后在临床研究的概念验证上, 针对BRCA发生突变的患者, 因而得以获得良好的治疗效果, 美国FDA也因此将尼拉帕尼列入快速通道进行优先审批。
(编者按)