 2021, Vol. 56
2021, Vol. 56



2. 中国药科大学药学院药物化学系, 江苏 南京 210009
2. Department of Medicinal Chemistry, School of Pharmacy, China Pharmaceutical University, Nanjing 210009, China
2020年, 新冠病毒席卷全球, 严重影响着全世界人民的生命健康与生活质量。这场突如其来的疫情警示我们需要时刻做好迎战全新疾病的准备, 其中新药的研发创制尤为重要。药物根据其研发的类型可分为首创性药物(first-in-class)和跟随性药物(me too, me better), 二者在新药创制的过程中都具有重要的意义。首创性药物的研究是生物学驱动的, 需要发现全新的作用机制与治疗靶点, 以解决无药可用的重大疾病, 通常需要从全新的候选靶标蛋白的发现开始, 历经靶标确证、分子筛选、成药性优化、靶标再确证等一系列过程。首创性药物研究的周期长、投入大、风险高, 但成功的首创性药物往往具有革命性的重大意义, 不仅能收获丰厚的经济回报, 也能开拓全新的药物研究领域, 是药物研究的风向标[1]。例如2015年上市的靶向于细胞周期依赖性蛋白激酶(CDK4/6)的首创性药物哌柏西利(palbociclib), 其研究从2000年发现结构母核到药物最终上市, 耗时十余年。更早的靶标发现研究可以追溯到1960年的细胞周期调控过程。基于这一调控机制, 人们直到1990年才确证了细胞周期依赖性蛋白激酶(CDKs)的重要生物学功能及其与恶性肿瘤间的联系。正因如此, “细胞周期的关键调控因子”也获得了2001年的诺贝尔生理或医学奖。坚实的基础研究是促成首创性药物成功的关键, 2005年辉瑞公司成功克服化合物的选择性和毒性问题, 研制出全球首个高选择性的CDK4/6抑制剂哌柏西利, 并继续历经10年的临床研究后上市, 成为了本领域的拓荒者。哌柏西利的有效性和安全性经受住了市场的考验, 取得了卓越的销售成绩。目前, 已有超过20项不同结构类型的CDK4/6抑制剂正在进行临床研究, 各大药企也争相仿制。跟随性药物通常是为了解决已有药物存在的某项缺陷或为了实现专利的突破, 由于不需要经历靶标的发现和确证过程, 往往是化学驱动的研究, 其研发周期短、投入少、风险较低, 例如今年获批上市的用于治疗成人急性偏头痛药物瑞美吉泮(rimegepant)就是以首创性药物替卡吉泮(telcagepant)的结构为基础, 通过充分研究其分子结构及化学特性, 在其原有结构上进行合理改造获得的具有完全自主知识产权且药效更优的药物分子。
全新靶标的发现是首创性药物研究的开始, 需要综合运用生物学和化学的各项技术手段, 具体的研究过程在早期的评述文章中已有提及[2, 3]。对于首创性药物而言, 风险最大、难度最高的工作莫过于靶标和疾病之间的因果性确证, 即如何充分地证明药物分子产生的治疗学效应是由于分子对靶标的直接调控而引起的, 这一过程也称为靶标的再确证。换言之, 真正有效的靶标不在于多而在于精, 首创性药物成功的首要条件是需要正确的靶标, 如何深刻理解靶点的生物学功能和成药性之间的联系(即靶标与疾病是否是强关联)是能否促成首创性药物成功的关键。如果没有“正确”的靶标, 即便药物化学家设计出再完美的分子, 最终也不能成为有效的药物。这意味着靶标与对应疾病之间的因果性验证需要长时间的沉淀与积累。然而, 一直以来人们为了更快地推进药物研发进程, 往往只关注分子作用于靶标后产生的生物学效应或其对于相应生物标志物的调控, 缺少对靶标调控和疾病之间关系的全面研究(即未能充分阐明靶标与疾病间的因果性), 最终未能成功。例如针对阿尔茨海默症(老年痴呆, AD)的BACE1 (β-分泌酶, 又名β-淀粉样前体蛋白裂解酶)抑制剂, 研究超过30余年, 从1992年发现BACE1靶标至今, 陆续已有20个BACE1抑制剂进入临床研究, 多个重磅药物临床Ⅱ期/Ⅲ期宣布无效, 至今无一上市, 因此仍未能证明BACE1靶标通过调控脑内Aβ多肽进而治疗AD的有效性。
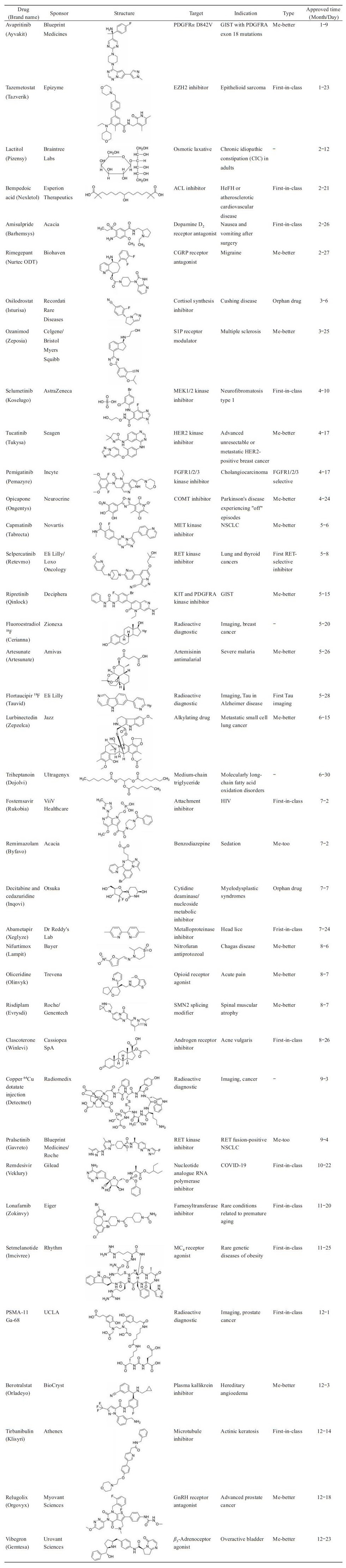
2020年, 在新冠疫情带来的巨大压力下, FDA共计批准上市新药53个, 仅次于2018年创纪录的59个。在2020年获批的新药中, 有38个小分子药物(总结于表 1)、2个RNA药物和13个生物药物。与最近几年趋势相同的是, 抗肿瘤药物依然占据主导, 今年获批的药物中包含18个抗肿瘤药物, 占比34%(在过去的5年中, 这一比例为25%)。紧随其后的是治疗神经类疾病的药物和抗感染类药物, 分别有8个和6个药物获批上市(占比分别为15%和11%)。近10年来, 孤儿药的获批数量持续增加, 今年获得孤儿药资格的药物为32个(占比60.4%), 仅次于2018年的34个。在今年获批的53款新药中, 23个属于first-in-class的首创性药物(占比达到40%), 包括首个酪氨酸蛋白激酶RET高选择性的单激酶抑制剂塞普替尼(selpercatinib), 用于治疗包括非小细胞肺癌、甲状腺髓样癌和其他类型的甲状腺癌在内的3种类型的肿瘤; 首款通过选择性抑制成纤维细胞生长因子受体(FGFR1/2/3)的胆管癌靶向药物佩米替尼(pemigatinib); 首个用于Tau蛋白的病理成像剂氟妥西吡[18F](flortaucipir 18F), 用于正在评估AD、伴有认知损害的成人患者, 确定脑部聚集性Tau神经纤维缠结(NFT)的密度和分布; 首个寡肽类黑素皮质素-4受体(MC4R)激动剂司美诺肽(setmelanotide), 可用于治疗前阿片黑素细胞皮质激素(POMC)和瘦素受体(LEPR)缺陷型肥胖症; 首款通过选择性抑制多巴胺D2和D3受体后用于对先前预防失败患者进行补救治疗的止吐药物氨磺必利(amisulpride); 另外还有多款首次获批的用于成像和诊断的试剂, 均为精准医疗发展提供了良好的保障。
| Table 1 Small molecule drugs approved by FDA in 2020.GIST:Gastrointestinal stromal tumor;ACL:Adenosine triphosphate-citrate lyase;MC4:Melanocortin 4;NSCLC:Non-small-cell lung cancer;UCLA:University California Los Angeles |
本文选取他泽司他(tazemetostat)、磷坦姆沙韦(fostemsavir)和氯那法尼(lonafarnib) 3个具有代表性的首创性小分子药物, 通过研发背景、药物设计思路及治疗学应用等方面简述其研究方法和研发过程, 以期为更多的首创性小分子药物提供借鉴与参考。
1 他泽司他(tazemetostat)——全球首个EZH2抑制剂用于治疗不适合手术的、转移性或局部晚期上皮样肉瘤 1.1 研发背景表观遗传领域的研究重点集中于翻译后修饰的过程(post-translational modification, PTMs), 其中组蛋白的甲基化扮演着关键角色[4]。在甲基化过程中, 赖氨酸和精氨酸是研究的重点, 基于其氨基酸的结构特征, 精氨酸可以被单/双甲基化, 而赖氨酸可以被单/双/三甲基化。不同的甲基化位点和甲基化程度往往会产生不同的生物学功能, 与相应的基因表达的激活、延伸和抑制密切相关, 其异常调控往往会导致恶性疾病的发生, 因此甲基化途径在靶向治疗中显示出了巨大的研究潜力[5]。
组蛋白的翻译后甲基化调控在基因组的调节中扮演重要的作用。其中, EZH2 (enhancer of zeste homolog2)是组蛋白甲基转移酶和多梳家族蛋白PRC2的催化亚单位, 其主要的生物学功能包括: 催化组蛋白H3的27位赖氨酸单甲基化、双甲基化和三甲基化。EZH2常见于多种免疫细胞并调控其发育、分化和生理功能。有趣的是, EZH2在血液肿瘤中表达量低, 但在肺癌、肉瘤等实体瘤中过度表达。EZH2的靶基因大多具有抑制肿瘤发生和调控干细胞分化的功能, 其过表达会导致相应的靶基因过度甲基化后功能沉默, 最终导致肿瘤的发生和干细胞的分化障碍。同时, EZH2的过表达通常会抑制T细胞的抗肿瘤反应, 增强肿瘤细胞的活力, 降低肿瘤抗原的表达, 导致T细胞趋化因子表达释放的降低, 从而使得肿瘤细胞产生对免疫细胞的耐受。因此, 基于EZH2在多种恶性肿瘤中的过表达现象及其作用机制, 使得靶向EZH2的药物研究成为可能。
上皮样肉瘤(epithelioid sarcoma, ES)是一种较为罕见的软组织肉瘤, 常见于青少年的皮下组织之中(在20~40岁的青壮年中男女患病比例为2∶1)。当前使用的治疗方法仍为传统的化疗手段, 疗效有限且不良反应大、患者生活质量较差, 在他泽司他获批前尚无有效的治疗上皮样肉瘤的方法。由于其多存在于软组织中, 一旦发生肿瘤的转移, 大多数患者的生存期难以超过一年。研究表明, 90%的上皮样肉瘤患者会伴有INI1蛋白的缺失, 此蛋白的缺失会导致EZH2的过度表达, 促使肿瘤细胞的恶性增殖。他泽司他的研究始于2000年初, 在历经十多年的基础研究后, 2011年3月Epizyme公司与卫材公司(Eisai)决定合作共同研发靶向EZH2的抗肿瘤药物, 又历经8年的临床前研究与临床研究, 他泽司他于2019年5月30日向FDA提交了上市申请并获得了加速审批和优先评审的快速通道, 最终成为全球首个靶向EZH2的首创性药物。
1.2 研发过程Epizyme公司首先通过基于多样性化学结构的高通量筛选寻找可以抑制EZH2的化学结构类型[6]。在最初的筛选过程中, 虽然发现的不少化合物都具有一定的EZH2酶活抑制能力, 但很多化合物因具有PAINS结构(pan assay interference compounds, 假阳性化合物)、氧化还原片段或易形成微粒聚集而被排除[7, 8]。真正的苗头化合物(hit)需要通过直接结合至EZH2而产生相应的抑制活性。最终, 通过多轮的筛选与反复验证, 得到了苗头化合物1 (IC50=3.4±0.9 μmol·L-1)[9]。如图 1所示, 苗头化合物1中含有4, 6-二甲基吡啶酮结构, 有趣的是这与同时期其他的四家公司筛选得到的化合物具有相同的母核结构, 也证实了这一结构特征对EZH2抑制活性的重要作用[10-12]。

|
Figure 1 Discovery and development of tazemetostat, from hit-to-lead to approved drug |
然而苗头化合物的活性及各项性质往往是不尽如人意的, 研究中发现化合物1不仅溶解性很差(在pH=7的中性条件溶解性小于10 μmol·L-1), 口服生物利用度也极低(F=0.5%)。为解决化合物1的难溶性问题, 研究人员首先希望通过引入极性基团来提高化合物的溶解性。基于苗头化合物1的结构特征, 通过继续筛选化合物1的相似物并研究其构效关系后成功发现了溶解性大幅提升、活性保持的先导化合物2(IC50=3.1±1.0 μmol·L-1), 完成了从hit-to-lead的优化过程。接下来的构效关系研究集中于对双环结构上的取代基替换, 结果表明苯并咪唑结构是最优的骨架核心, 同时研究发现吡啶N1上的取代基对活性影响较大, 且活性随着取代基体积的增大而增加, 最终环戊基取代的化合物活性最优(化合物3, IC50=2.9±1 μmol·L-1)。通过对化合物结构的深入分析, 研究人员发现苯并咪唑的平面结构并不是最优的结合模式, 因此尝试打破这一双环结构并发现了化合物4 (IC50=0.7±0.2 μmol·L-1), 其抑制活性进一步提升。有趣的是, 对亚胺结构上烷基的取代研究表明, 乙基取代后的产物抑制活性更强。将化合物4中的环戊基替换为四氢吡喃环后, 溶解度和活性进一步提升, 最终发现的化合物5即他泽司他, 表现出更强效的抑制活性(IC50=0.2±0.1 μmol·L-1)、更低的体内清除率(CL=16 mL·min-1·kg-1)和更优的生物利用度(F=55%)[9]。
1.3 治疗应用本文中列举的他泽司他是全球第一个获批的EZH2小分子抑制剂, 他泽司他通过阻断EZH2的甲基转移酶活性, 有效阻止癌细胞生长, 用于治疗16岁及以上患有转移性或局部晚期不可切除的上皮样肉瘤患者。除了上皮样肉瘤外, 基于EZH2的作用机制和调控特点, 他泽司他目前有超过20项的临床研究正在进行, 包括前列腺癌(NCT04179864)、头颈部鳞癌(NCT04624113)、淋巴瘤(NCT02875548)、间皮瘤(NCT02860286)等。除了他泽司他, 正在进行临床研究的EZH2抑制剂还包括GSK公司的GSK2816126、Constellation Pharmaceuticals的CPI-1205和恒瑞医药的SHR2554等。
2 磷坦姆沙韦(fostemsavir)——全球首个新型附着抑制剂用于治疗HIV-1感染 2.1 研发背景人类免疫缺陷病毒(HIV)主要包括两种类型: HIV-1和HIV-2, 其中HIV-1的毒性和传染性更强。HIV主要通过血液和体液传播, 破坏人体的CD4 T细胞, 进而损害整个人体的免疫系统。目前, HIV的治疗药物主要包括核苷类和核苷酸类反转录酶抑制剂(NRTIs)、非核苷类反转录酶抑制剂(NNRTIs)、蛋白酶抑制剂(PIs)、整合酶抑制剂(INSTIs)和融合抑制剂(FIs)。尽管现有的药物及其组合疗法已经能很大程度上控制病毒对人类的伤害(在美国, 死亡人数从2005年的190万降至2016年100万), 使得艾滋病(AIDS)逐渐变成一种慢性病, 但不断创新高的感染人数仍是HIV-1病毒对人类的巨大威胁。不仅如此, 由于HIV病毒具有不断变化的能力, 尽管在过去的30年里HIV的治疗药物已取得了突飞猛进的成绩, 但仍然可以发现一些患者会对抗逆转录病毒药物产生耐药性, 导致既往的治疗方案失败。对于接受过多种既往治疗方案但产生多重耐药的患者群体而言, 磷坦姆沙韦的出现使其医疗需求得到了满足, 将成为此类HIV患者的重要的治疗选择[13, 14]。
2.2 研发过程磷坦姆沙韦的相关研究可追溯至将近20年前, 百时美施贵宝(BMS)公司的研究人员于2003年发现的苗头化合物6[15]。如图 2所示, 通过一项基于表型的细胞水平筛选实验, 研究人员获得了一类可以附着于HIV-1病毒gp120糖蛋白的吲哚-3-乙二醛胺类化合物, 通过全新的作用机制抑制HIV-1。虽然苗头化合物6已表现出较强的抗病毒活性, 但较强的毒性限制了其进一步的研究。随后的构效关系着眼于优化此类化合物的吲哚环结构, 研究过程中发现吲哚环结构中的4位引入甲氧基后可以大幅提升活性, NH位也可进行不同程度的取代, 7位有取代基时活性也会有所提高, 基于以上的初步研究结果得到了6-氮杂吲哚类衍生物7 (BMS-488043, EC50=0.88±0.46 nmol·L-1), 活性提升的同时不良反应也大幅下降。临床试验结果表明, 尽管化合物7通过新型的附着机制展现了一定的疗效, 但其药效响应仍不理想(以超过1 lg copies/mL为标准, 800 mg剂量组响应率58%, 1 600 mg剂量组响应率67%)。基于化合物7的结构, 研究人员又进行了更为全面的构效关系研究, 最终得到了化合物8 (替米沙韦, temsavir)。化合物8是通过对化合物7上的C7位进行取代基的替换研究后发现的, 表现出了更强的活性(EC50=0.14 nmol·L-1)和更优的药代动力学(PK)性质, 是目前最优的HIV-1附着抑制剂。研究人员通过将其制成前药9 (磷坦姆沙韦)以克服药物因为溶解性差而导致的吸收问题, 获得了更好血浆暴露量和更优的体内药效[16, 17]。

|
Figure 2 Discovery and development of fostemsavir as a prodrug of temsavir |
磷坦姆沙韦是一种首创性的HIV-1附着抑制剂, 获得了FDA授予的快速通道资格和突破性药物资格。基于其独特的作用机制, 磷坦姆沙韦可与其他抗逆转录药物联用, 治疗多重耐药或因耐药性和安全性等问题接受过大量现有方案但治疗失败的成人HIV-1感染者[18]。正是因为不断地有全新机制的抗HIV药物被研发出来, HIV再也不是“不治之症”, 其患者可以有更多治疗的选择并获得更长时间的健康。
3 氯那法尼(lonafarnib)——全球首个异戊二烯化抑制剂用于治疗早衰症 3.1 研发背景哈金森-吉尔福德早衰综合征(HGPS, 简称早衰症)和早老样核纤层蛋白病(progeroid lami‐nopathies)是非常罕见的遗传疾病, 患病者常见于儿童, 主要表现为衰老速度显著加快。早衰症在氯那法尼获批前无有效治疗手段。早衰症的发病原因主要是因为LMNA基因(核纤层蛋白家族)突变而导致的异戊二烯化异常, 使核膜蛋白A (lamin A)过度积累而导致的。核膜蛋白A是细胞核结构和功能的重要组成部分, 突变的LMNA基因会生成异常的核膜蛋白A, 这种异常蛋白缺少去除经过异戊二烯化修饰C端蛋白片段的能力。长时间后, 异常的核膜蛋白A会在细胞核内膜积累, 影响细胞核的稳定性, 最终导致早衰症的发生。早衰症患者如未及时接受治疗, 平均寿命仅有14.5年。法呢基转移酶(FTase)是一种参与异戊二烯化修饰过程的蛋白酶, 通过有效靶向于法呢基转移酶可以实现对核膜蛋白A异戊二烯化的抑制, 以此降低核膜蛋白A在细胞核中的过渡积累, 实现治疗效果。
3.2 研发过程针对法呢基转移酶的研究始于多肽类抑制剂, 由一类CAAX (C: 半胱氨酸, A: 任意脂肪族氨基酸; X: 任何氨基酸)的四肽发展而来[19]。随着对此类靶标认知的不断深入, 由Schering-Plough研究所的研究人员筛选并发现了一类具有全新骨架类型的法呢基转移酶抑制剂(如图 3所示), 化合物10 (IC50=27 000 nmol·L-1)是其最早发现的具有一定抑制活性的苗头化合物, 其结构是H1受体拮抗剂氯雷他定的类似物。随后通过构效关系研究得到的化合物11(IC50=250 nmol·L-1), 活性大幅提升, 其对法呢基转移酶的抑制活性提高了100倍。在三环结构的吡啶3位引入溴原子后, 不仅可以提升活性, 也进一步提高了化合物的PK性质, 在此基础上得到的化合物12不仅具有更强的抑制活性(IC50=90 nmol·L-1), 也实现了口服的有效性[20]。接下来的研究发现通过替换化合物12中的吡啶氮氧化物, 得到的化合物13的活性又进一步提升(IC50=49 nmol·L-1)。基于13的结构在其三环的苯环上引入溴原子后得到的化合物14 (SCH-66336), 即为氯那法尼(IC50=1.9 nmol·L-1)[21, 22]。有趣的是, 氯那法尼最早的治疗应用为抗肿瘤, 并对多种人类的肿瘤细胞有较好的抗增殖抑制活性[23]。

|
Figure 3 Discovery and development of lonafarnib |
氯那法尼是一种口服有效的法呢基转移酶抑制剂, 临床试验数据表明接受氯那法尼单药治疗后的早衰症儿童患者的死亡风险降低88%。氯那法尼基于其全新的作用机制与靶标, 被FDA和欧洲药品管理局(EMA)均指定为孤儿药, 并获得了突破性疗法认证。
有趣的是, 除了治疗早衰症, 氯那法尼同时还获得了治疗丁型肝炎(HDV)的突破性疗法认证。丁型肝炎是严重影响人类生命健康的病毒性肝炎, 相比其他肝炎类型, HDV感染后的病情更为严重, 尚无有效的治疗药物。丁型肝炎是由HDV病毒引起的, 仅在携带乙型肝炎病毒(HBV)的个体中作为共感染而发生, 因此常会引发比乙肝更严重的肝脏疾病, 并伴随有肝纤维化加速、肝癌和肝功能衰竭。法呢基转移酶是通过异戊二烯化过程调控的蛋白质的酶, HDV可利用宿主细胞内的这种重要酶类完成其生命周期的重要过程。因此, 抑制肝细胞中HDV病毒复制过程的异戊二烯化步骤, 可有效阻止病毒的繁殖。不仅如此, 还有多项研究结果表明氯那法尼可以在多种肿瘤表型中表现出优异的抗增殖效果[23-25]。
4 结语与展望除了以上浅析的首创性药物之外, 今年获批的药物中也有不少的特殊性。其中, “人民的希望”瑞德西韦是吉利德公司研发的首个用于治疗COVID-19的小分子药物, 其前所未有的获批速度也震惊了全世界。值得一提的是, 瑞德西韦作为一款RNA聚合酶抑制剂, 最早开发其用于丙型肝炎和呼吸道合胞体病毒的治疗, 但却在2014~2016年紧急作为在治疗埃博拉病毒的药物。虽然瑞德西韦在埃博拉病毒的治疗中未表现出良好的疗效, 但却提供了大量的安全性数据。在今年的新冠疫情中, 瑞德西韦的临床治疗于2月开始, 历经8个月的临床评价, 最终于10月上市。尽管最终的疗效饱受诟病, 但瑞德西韦在特殊时期也具有重要的意义。除以之外, 青蒿琥酯作为青蒿素类的第一个抗疟疾药物, 拯救了数万人的生命。青蒿琥酯是我国著名科学家刘旭教授在70年代攻坚克难研制成功的, 迄今为止挽救了超过2 400万重症疟疾患者的生命。虽然今年FDA的批准姗姗来迟, 但其作为治疗严重疟疾的首选药物仍彰显了我国科学家的首创性。青蒿琥酯的发明人刘旭教授于2019年辞世, 值得我们永远铭记。
在2020年FDA批准的新药中, 32%的药物获得了快速审批通道, 42%的药物获得了突破性疗法认证, 57%的药物获得了优先评审资格, 23%的药物获得了加速审批资格, 体现出了FDA对创新药开发和批准的速度与效率。随着我国监管改革的深化和推进, 中国的创新药企也正在逐渐缩小与全球高水平药企之间的差距。中国在2020年全年获批新药13个, 其中有10款是国内自主研发的创新产品。例如豪森药业的阿美替尼, 是全球第二个获批上市的三代EGFR-TKI药物; 百济神州的泽布替尼, 是中国首款获批的BTK抑制剂; 贝达药业的恩沙替尼, 打破了国内ALK阳性非小细胞肺癌靶向治疗均为进口药的垄断局面; 恒瑞的氟唑帕利, 是国内首个自主研发的PARP抑制剂等。随着我国国力和科研水平的不断提高, 相信在不久的将来会有更多的首创性分子会成为“中国制造”。
作者贡献: 王磊负责文章资料收集与撰写; 尤启冬负责文章的选题与修改, 为该文章的主要负责人。
利益冲突: 本文无利益冲突。
| [1] |
Guo ZR. Concise analysis for innovation of pioneering and follow-on drugs[J]. Acta Pharm Sin(药学学报), 2016, 51: 1179-1184. |
| [2] |
Wang L, Jiang ZY, You QD. First-in-class small molecule drugs in 2018[J]. Acta Pharm Sin(药学学报), 2018, 54: 1145-1156. |
| [3] |
Wang L, You QD. First-in-class small molecule drugs in 2019[J]. Acta Pharm Sin(药学学报), 2020, 55: 1983-1994. |
| [4] |
Barreiro EJ, Kummerle AE, Fraga CA. The methylation effect in medicinal chemistry[J]. Chem Rev, 2011, 111: 5215-5246. DOI:10.1021/cr200060g |
| [5] |
Leung CS, Leung SS, Tirado-Rives J, et al. Methyl effects on protein-ligand binding[J]. J Med Chem, 2012, 55: 4489-4500. DOI:10.1021/jm3003697 |
| [6] |
Knutson SK, Wigle TJ, Warholic NM, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells[J]. Nat Chem Biol, 2012, 8: 890-896. DOI:10.1038/nchembio.1084 |
| [7] |
Lor LA, Schneck J, Mcnulty DE, et al. A simple assay for detection of small-molecule redox activity[J]. J Biomol Screen, 2007, 12: 881-890. DOI:10.1177/1087057107304113 |
| [8] |
Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds(PAINS)from screening libraries and for their exclusion in bioassays[J]. J Med Chem, 2010, 53: 2719-2740. DOI:10.1021/jm901137j |
| [9] |
Kuntz KW, Campbell JE, Keilhack H, et al. The importance of being me: magic methyls, methyltransferase inhibitors, and the discovery of tazemetostat[J]. J Med Chem, 2016, 59: 1556-1564. DOI:10.1021/acs.jmedchem.5b01501 |
| [10] |
Verma SK, Tian XR, LaFrance LV, et al. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2[J]. ACS Med ChemLett, 2012, 3: 1091-1096. DOI:10.1021/ml3003346 |
| [11] |
Gehling VS, Vaswani RG, Nasveschuk CG, et al. Discovery, design, and synthesis of indole-based EZH2 inhibitors[J]. Bioorg Med ChemLett, 2015, 25: 3644-3649. DOI:10.1016/j.bmcl.2015.06.056 |
| [12] |
Qi W, Chan H, Teng L, et al. Selective inhibition of EZH2 by a small molecule inhibitor blocks tumor cells proliferation[J]. Proc Natl Acad Sci U S A, 2012, 109: 21360-21365. DOI:10.1073/pnas.1210371110 |
| [13] |
Beyrer C, Pozniak A. HIV drug resistance-an emerging threat to epidemic control[J]. N Engl J Med, 2017, 377: 1605-1607. DOI:10.1056/NEJMp1710608 |
| [14] |
Wainberg MA, Zaharatos GJ, Brenner BG. Development of antiretroviral drug resistance[J]. N Engl J Med, 2011, 365: 637-646. DOI:10.1056/NEJMra1004180 |
| [15] |
Wang T, Zhang Z, Wallace OB, et al. Discovery of 4-benzoyl-1-[(4-methoxy-1H-pyrrolo[2, 3-b]pyridin-3-yl)oxoacetyl]-2-(R)-methylpiperazine(BMS-378806): a novel HIV-1 attachment inhibitor that interferes with CD4-gp120 interactions[J]. J Med Chem, 2003, 46: 4236-4239. DOI:10.1021/jm034082o |
| [16] |
Meanwell NA, Krystal MR, Nowicka-Sans B, et al. Inhibitors of HIV-1 attachment: the discovery and development of temsavir and its prodrug fostemsavir[J]. J Med Chem, 2018, 61: 62-80. DOI:10.1021/acs.jmedchem.7b01337 |
| [17] |
Wang T, Ueda Y, Zhang ZX, et al. Discovery of the human immunodeficiency virus type 1(HIV-1)attachment inhibitor temsavir and its phosphonooxymethyl prodrug fostemsavir[J]. J Med Chem, 2018, 61: 6308-6327. DOI:10.1021/acs.jmedchem.8b00759 |
| [18] |
Thompson M, Lalezari JP, Kaplan R, et al. Safety and efficacy of the HIV-1 attachment inhibitor prodrug fostemsavir in antiretroviral-experienced subjects: week 48 analysis of AI438011, a Phase IIb, randomized controlled trial[J]. Antivir Ther, 2017, 22: 215-223. |
| [19] |
Bell IM. Inhibitors of farnesyltransferase: a rational approach to cancer chemotherapy?[J]. J Med Chem, 2004, 47: 1869-1878. DOI:10.1021/jm0305467 |
| [20] |
Njoroge FG, Vibulbhan B, Rane DF, et al. Structure-activity relationship of 3-substituted N-(pyridinylacetyl)-4-(8-chloro-5, 6-dihydro-11H-benzo[5, 6]cyclohepta[1, 2-b]pyridin-11-ylidene)-piperidine inhibitors of farnesyl-protein transferase: design and synthesis of in vivo active antitumor compounds[J]. J Med Chem, 1997, 40: 4290-4301. DOI:10.1021/jm970464g |
| [21] |
Njoroge FG, Taveras AG, Kelly J, et al. (+)-4-[2-[4-(8-Chloro-3, 10-dibromo-6, 11-dihydro-5H-benzo[5, 6]cyclohepta[1, 2-b]-pyridin-11(R)-yl)-1-piperidinyl]-2-oxo-ethyl]-1-piperidinecarboxamide(SCH-66336): a very potent farnesyl protein transferase inhibitor as a novel antitumor agent[J]. J Med Chem, 1998, 41: 4890-4902. DOI:10.1021/jm980462b |
| [22] |
Taveras AG, Aki C, Chao J, et al. Exploring the role of bromine at C(10)of(+)-4-[2-[4-(8-chloro-3, 10-dibromo-6, 11-dihydro-5H-benzo[5, 6]cyclohepta[1, 2-b]pyridin-11(R)-yl)-1-piperidinyl]-2-oxoethyl]-1-piperidinecarboxamide(Sch-66336): the discovery of indolocycloheptapyridine inhibitors of farnesyl protein transferase[J]. J Med Chem, 2002, 45: 3854-3864. DOI:10.1021/jm010463v |
| [23] |
Liu M, Bryant MS, Chen J, et al. Antitumor activity of SCH66336, an orally bioavailable tricyclic inhibitor of farnesyl protein transferase, in human tumor xenograft models and wap-ras transgenic mice[J]. Cancer Res, 1998, 58: 4947-4956. |
| [24] |
Caponigro F. Farnesyl transferase inhibitors: a major breakthrough in anticancer therapy? Naples, 12 April 2002[J]. Anticancer Drugs, 2002, 13: 891-897. DOI:10.1097/00001813-200209000-00016 |
| [25] |
Winquist E, Moore MJ, Chi KN, et al. A multinomial Phase II study of lonafarnib(SCH 66336)in patients with refractory urothelial cancer[J]. Urol Oncol, 2005, 23: 143-149. DOI:10.1016/j.urolonc.2004.12.012 |