 2021, Vol. 56
2021, Vol. 56

间变性淋巴瘤激酶(anaplastic lymphoma kinase, ALK) 属于胰岛素受体家族的酪氨酸激酶, 2007年发现在肺癌患者中由于染色体倒位, 形成棘皮动物微管相关类蛋白4 (EML4) 基因和ALK基因的重排(EML4-ALK), 促使肺癌发生和发展。EML4-ALK基因重排(又称ALK阳性, ALK+) 的肺癌是新近发现的一种分子亚型, 主要发生在非小细胞肺癌, 约占肺癌3%~5%。第一个上市治疗ALK+的非小细胞肺癌靶向药物是克唑替尼(1, crizotinib), 患者对克唑替尼的应答率约70%, 无进展生存8~11个月。用药后由于ALK发生变异, 而产生耐药性。第二代靶向药物是针对I1171N/T/S变异的埃雷替尼(2, alectinib)、针对F1174C/V和G1202R变异的色瑞替尼(3, ceritinib)。第三代药物的研究目标是对各种变异的ALK+非小细胞肺癌都有效的药物。
2 含氧化二甲膦片段的先导物严格意义上讲, 研发第三代药物不属于首创性, 但也不会是跟随性的模仿, 因为要对耐药的靶标有效, 结构上一定有特色, 何况作用于ALK激酶的已上市的、处于临床和正在临床前的抑制剂有多种结构骨架, 不能侵犯知识产权。
|
ARIAD公司一开始探索了多种结构, 其中一个是以多见的2-苯胺基嘧啶为母核, 在苯环的2'位连接甲氧基, 目的是使甲氧基结合于ALK酶的铰链附近的小疏水腔, 以避免对其他激酶的结合(减少分子的杂泛性)。最重要的是在4位引入氧化二甲基膦片段(DMPO), DMPO是氢键接受体, 结构新颖, ARIAD公司曾研究Src激酶抑制剂, 有过引入DMPO基团提高活性70倍的经验(Dalgarno D, Stehle T, Narula S, et al. Structural basis of Src tyrosine kinase inhibition with a new class of potent and selective trisubstituted purine-based compounds. Chem Biol Drug Des, 2006, 67: 46-57)。嘧啶4位连接取代的苯胺片段, 作为优化的位点, 所以设计的先导物是含有DMPO的2, 4-二苯胺基嘧啶结构骨架。
在评价化合物对ALK的抑制活性(IC50) 的同时测定对胰岛素样生长因子受体(IGF1R) 和胰岛素受体激酶(InsR) 的抑制作用(IC50), 因为IGF1R和InsR与ALK的蛋白序列同源性较高, 为避免影响血糖的杂泛性, 评价化合物的脱靶作用, 显示ALK的选择性。在细胞水平上评价化合物对ALK+的Karpas299细胞和对ALK-的U937细胞的抑制作用(IC50), 二者的活性差异显示化合物的选择性。首轮合成的化合物列于表 1。
| 表 1 Activity of compounds synthesed in the first round |

表 1的构效关系如下: ① C2连接的苯胺基的4'-DMPO对活性为正贡献, 因为化合物10与4的区别在于10没有4'-DMPO基团, 活性降低7倍。② C4连接的苯胺基上的2'-取代(4~9) 保持了高活性, 这些基团都是氢键接受体。③化合物对ALK+细胞的活性显著高于ALK-细胞, 提示对ALK+有选择性作用。④化合物4的异丙磺酰基是借鉴色瑞替尼(3) 的片段, 对ALK-细胞活性很高(IC50 = 20 nmol·L-1), 但对IGF1R的抑制活性接近于ALK, 选择性差, 因而未作为优选的基团。⑤化合物9在4-苯胺环上也有DMPO, 虽然细胞水平的活性不高, 但对ALK的选择性显著强于其他化合物。为此固定4-苯胺基的2'-DMPO, 下一步变换C2-苯胺基环上的基团。
3 C2-苯胺基环上的变换C4连接的苯胺环的邻位固定为氧化二甲膦基, 考察在C2连接的苯胺环的对位引出碱性基团, 一是提高对ALK的活性, 特别是ALK+细胞活性, 另一考虑是该位置接近酶的边沿, 可以提高分子的溶解性。因而合成了含有不同碱性基团的集中库, 列于表 2中。
| 表 2 Activity of compounds that change the substituents on the benzene ring attached to C2 |

表 2化合物对酶和细胞活性的构效关系分析如下: ①所有的化合物对ALK酶显示强抑制活性, 变换环的结构对抑酶活性没有显著差异, 但对细胞活性有区别, 推测是由于细胞内ATP浓度较高, 温孵72 h显示出差异, 也可能是由于细胞为全长的ALK激酶, 而测定酶活性的是部分结构。②直接与苯环相连的哌嗪或哌啶化合物如15和19有较高细胞活性, 环末端被氧或碳原子置换或降低氮的碱性则活性降低, 例如14、16、17和18。③含氮的七元或五元环细胞活性低于六元环。④化合物30是移植克唑替尼的助溶基团(跟进性药物研究惯用方法), 但活性很差, 可能是芳环吡唑不利于细胞内的结合。⑤化合物15、23、24、27和31对ALK+细胞有较高活性和选择性(相对于野生型和胰岛素受体), 27的选择性尤为突出。
4 嘧啶环C5、C2与C4苯环上取代基的变换以化合物27为中心, 对C5和C2与C4的取代基作微调变换, 优化对细胞的活性和选择性, 以求精准化设计, 合成的化合物列于表 3。
| 表 3 Activity of the compounds of different substituents on pyrimidine ring C5, and benzene ring C2 and C4 |

变换各个位置的基团与活性变化的关系如下: ①嘧啶环的C5连接的氯原子被其他基团或原子取代(32~35) 都使细胞活性降低, 尤其是乙酰基置换(36) 活性殆尽, 所以C5为氯原子是最佳基团。② C4连接的苯胺环上的2''-氧化二甲膦基换成二乙基, 化合物37对细胞的抑制降低5倍, 提示结合腔的体积不能容纳较大的二乙基氧膦。③ C2连接的苯胺环上2'甲氧基换成乙氧基(40) 或异丙氧基(41) 虽然降低了对胰岛素受体的作用, 但对ALK和细胞的抑制降低了50%, 置换得不偿失。④ 4'位被F取代(38) 显著降低细胞活性, 而甲基取代(39) 的细胞活性略升, 但酶活性略降。
综上, 化合物27仍是优良的化合物。为了证明氧化二甲膦是该项目的特色结构, 进而换成其他氢键接受体以确定DMPO的必要性。表 4列出了变换C4苯胺环上2''的基团的化合物及其活性。
| 表 4 Activity of the compounds (change the dimethylphosphine oxide to other substituent) (IC50/nmol·L-1) |
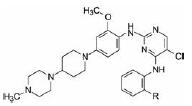
分析表中的构效关系如下: ①氰基(44) 和羟乙基(50) 化合物对胰岛素受体的作用虽然较弱, 但抑制ALK的活性也降低了3~5倍。②乙酰基、磺酰胺基和磺酰基等氢键接受体, 对ALK和胰岛素受体选择性较低, 与基团的尺寸大小无关, 说明二甲基膦氧基团对于活性和选择性是不可替换的。化合物52再一次借鉴色瑞替尼的成功基团, 依然没有成功。
5 确定候选化合物和布加替尼上市在上述诸轮优化的过程中, 对活性和选择性较强的化合物, 进行了大鼠和小鼠的药代动力学评价, 在第3节优化C2连接的苯环4'位的碱性基团出现了不少高活性化合物, 但选择27作为里程碑式化合物作精准性优化, 是因为N-甲基哌嗪基哌啶片段的药代性质优于其他环系, 例如四氢吡咯哌啶、羟乙基哌嗪等化合物的清除率很高(数据从略), 较低的生物利用度预示着难以成药的风险。
用Karpas-299小鼠模型评价高活性化合物的药代(PK) 和药效(PD) 性质, 小鼠灌胃25 mg·kg-1, 在10、16和24 h测定细胞中ALK被磷酸化的百分率(与空白对照比, 越低越好)。表 5列出的数据表明, 化合物15和27呈现稳定的抑制磷酸化性质, 尤其是27在灌胃16和24 h只有7%和39%被磷酸化, 提示持续性抑制作用, 这个PK/PD实验预示体内有较好的抗肿瘤效果。
| 表 5 Percentage of ALK phosphorylation at different time of compounds (25 mg·kg-1, intragastric administration) |
进而评价化合物15、17、24和27对Karpas-299小鼠的抗肿瘤活性, 同等剂量下27显著优于其他化合物(数据从略), 从而作为候选化合物, 定名为布加替尼(brigatinib), 经临床前和临床研究, 表明对克唑替尼治疗后病情进展的, 或对克唑替尼不能耐受的ALK阳性的局部晚期或转移性非小细胞肺癌是有效的药物。由于疗效显著, 美国FDA加速批准, 于2017年4月28日批准上市(Huang WS, Liu SY, Zou D, et al. Discovery of brigatinib (AP26113), a phosphine oxide-containing, potent, orally active inhibitor of anaplastic lymphoma kinase. J Med Chem, 2016, 59: 4948-4964)。

|
X射线分析布加替尼与ALK复合物晶体结构表明(PDB 5J7H), 布加替尼分子呈U形构象定位于ATP结合位点, 嘧啶环位于腺苷结合处, 甲氧基邻近于铰链, 与L1198相互作用, 氯原子与门户残基L1196结合, 带有二甲膦氧的苯胺环处于DFG部位, P=O与苯胺的NH发生分子内氢键结合, 稳定了U形药效构象。甲基哌嗪处于无结合的自由状态, 发挥助溶作用(图 1)。

|
图 1 The crystal structure of the complex of brigatinib and ALK, the dotted line represents the hydrogen bond |
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
精准医学需要有准确的诊断, 使用精确设计研发的药物。布加替尼的研制体现了精准的要求, 不仅针对ALK阳性的非小细胞肺癌, 而且是对克唑替尼治疗后耐药的患者。虽然在临床前的研究中没有直接针对克唑替尼耐药的酶和细胞, 但从先导物的设计和评价开始, 就重视酶和细胞水平的选择性作用。在化学上, 含有机膦基团是布加替尼的结构特色, 加之其他基团的精细优化, 成就了临床研究的既定目标。由于显著疗效, FDA曾确定布加替尼为突破性资格药物、孤儿药资格, 并在2017年4月28日, 加速批准了布加替尼在美国上市。
(编者按)