 2021, Vol. 56
2021, Vol. 56



抑郁症是世界范围内导致残疾的主要原因之一, 极大地加剧了全球疾病负担。目前, 全世界有超过3.4亿人受到抑郁症的困扰[1]。抑郁症临床表现为持续的心境低落、兴趣丧失以及精力缺乏; 常伴有其他认知、生理及行为的异常如注意力下降、体重及食欲的变化和失眠等, 严重的抑郁症甚至可能导致自残、自杀行为。抑郁症可以影响任何年龄段, 并且可以在整个生命周期中持续存在或复发。由于青春期、围产期和更年期相关的抑郁风险增加, 因而女性患抑郁症的可能性比男性高约50%[2]。目前, 抑郁症主要采取心理治疗、物理治疗和药物治疗[3], 后者是中重度抑郁症的一线疗法。抗抑郁药主要包括五羟色胺再摄取抑制剂(serotonin reuptake inhibitors, SSRI)、五羟色胺及去甲肾上腺素再摄取抑制剂(serotonin and norepinephrine reuptake inhibitors, SNRI)和三环类抗抑郁药(tricyclic antidepressant, TCA)等, 但这些药物起效慢、不良反应多且治疗效果有限, 研究显示, 药物治疗下约三分之二的重症抑郁患者未能缓解[4]。近年来, 随着对抑郁症发病机制研究的深入, 出现了非传统单胺递质系统的新型抗抑郁症药物, 如2011年美国食品与药品管理局(Food and Drug Administration, FDA)批准了靶向褪黑素和五羟色胺受体的阿戈美拉汀(agomelatine), 2019年FDA批准一种谷氨酸N-甲基-D-天冬氨酸(glutamate N-methyl-D-aspartate, NMDA)受体拮抗剂——艾司氯胺酮(esketamine)用于治疗难治性抑郁症(treatment resistant depression, TRD)[5]。然而, 这些新型药物仍然存在因靶点在体内广泛分布而带来的不良反应, 以及对神经系统的潜在毒性和成瘾性等安全问题, 因此, 对临床抗抑郁药物的研究仍需要进一步深入。
抑郁症是在环境、遗传、生理和心理因素共同作用下导致的心理疾病。全基因组关联研究确定了某些与抑郁症发病风险相关联的基因[6], 如Elav样RNA结合蛋白2 (Elav like RNA binding protein 2, ELAVL2)、CUGBP Elav样家族成员4 (CUGBP Elav-like family member 4, CELF4)和神经生长调节蛋白1 (neuronal growth regulator 1, NEGR1)等, 这些基因与突触功能及活性关系密切, 其中ELAVL2也参与神经发育[7]。另一方面, 越来越多的证据表明个体对抑郁的敏感性受到遗传和环境系统之间存在的复杂相互作用的影响。在此过程中, 表观遗传学(epigenetic)可能是一种关键的调控机制: 表观遗传学是指在不改变脱氧核糖核酸(deoxyribonucleic acid, DNA)序列的情况下改变基因表达的可遗传变化, 包括DNA甲基化、组蛋白(histone)修饰、核小体重塑和非编码RNA表达等[8]。这一过程可以被多种因素激活, 例如个体早期的成长环境、药物使用、衰老和饮食等[9]。研究认为, 机体遭受应激如儿童时期遭遇创伤或生活中承受严重压力时, 神经元可能通过DNA甲基化或组蛋白修饰调节突触可塑性相关的基因, 从而对各种环境信号作出反应[10], 当机体作出失调的适应性反应时, 则可能导致抑郁的发生[11]。
大量研究表明, 组蛋白修饰在包括抑郁症、焦虑症、恐惧症和精神分裂症在内的精神疾病中发挥重要作用[12, 13]。染色质由DNA和组蛋白等组成, 其基本结构单位是核小体, 核小体由各两分子的4种不同组蛋白(组蛋白H2A、H2B、H3和H4)组成八聚体[14]。组蛋白修饰通过改变细胞核中染色质的构象或募集重构酶, 利用来自ATP水解的能量重新定位核小体, 可正向或负向调节基因的转录[15]。目前发现的组蛋白翻译后修饰(histone post-translational modifications, PTMs)包括甲基化、乙酰化、磷酸化、泛素化、丙二酰化、丁酰化和巴豆酰化[16]等。其中乙酰化是组蛋白最广泛的修饰之一, 其修饰程度受机体代谢状态的精细调控。常见修饰有H4K12ac (表示H4组蛋白的第12位赖氨酸发生乙酰化, 下同)、H3K12ac等。组蛋白乙酰基转移酶(histone acetyltransferases, HATs)催化乙酰化过程, 使从核小体伸出的N-末端组蛋白尾部的赖氨酸残基带负电荷, 这些负电荷会排斥带同种电荷的DNA, 导致染色质结构松弛, 使其处于更加开放的状态, 允许转录因子结合并显著增加基因表达[16], 这一过程可由组蛋白去乙酰化酶(histone deacetylases, HDAC)逆转。乙酰化修饰对蛋白质功能具有重要调节作用, 参与基因转录、蛋白降解和细胞代谢等多个细胞过程[17, 18]。
1 HDAC与抑郁症 1.1 HDAC的类型目前已知有18种HDAC, 根据结构、催化功能和细胞定位的不同可分为两个家族: 依赖Zn2+的HDAC(Zn2+-dependent HDAC)和依赖NAD+的沉默调节蛋白(NAD+-dependent sirtuins); 或者4个类别: I、IIa/IIb、III和IV, 其中I、IIa/IIb、IV类属于依赖Zn2+的HDAC, III类属于依赖NAD+的沉默调节蛋白。HDAC在体内广泛表达, 发挥修饰组蛋白和非组蛋白、调节染色质结构、抑制或激活基因表达以及调节细胞代谢的作用[13]。
I类HDAC (HDAC I: HDAC1、2、3和8)与组蛋白去乙酰化酶1 (histone deacetylase 1, HDA1)基因有同源性。HDAC I在体内表达广泛, 主要定位于细胞核, 并对底物有较高的酶促活性[19]。
II类HDAC (HDAC IIa: HDAC4、5、7和9;HDAC IIb: HDAC6和10)与HDA1基因有同源性[20]。HDAC IIa位于细胞核中, 一般情况下处于非磷酸化状态, 响应特定信号时被磷酸化, 与转录因子相互作用而被募集到靶基因, 发挥抑制其转录的作用[21]。HDAC IIb具有2个脱乙酰基酶结构域, 参与微管和肌动蛋白依赖性细胞运动的调控[22]。
III类HDAC (HDAC III)也称为sirtuins (SIRT)包括SIRT1~7, 是一类依赖NAD+的沉默调节蛋白[23]。SIRT存在于细胞核、细胞质和线粒体中, 执行调控细胞存活、代谢、凋亡等生理功能[24]。
IV类(HDAC IV)仅包含1个酶: HDAC11。HDAC11在大脑、心脏、肌肉、肾脏和睾丸都有丰富表达, 然而目前研究者对其功能知之甚少[25]。
1.2 HDAC与抑郁症病理的关系 1.2.1 临床试验Hobara等[26]比较了20名未经治疗的重症抑郁(major depression disorder, MDD)患者和28名健康志愿者的外周血白细胞中HDAC1~11的mRNA水平, 发现MDD患者的抑郁发作期(而非缓解期)外周血白细胞中HDAC2和HDAC5 mRNA的水平增加。Ige等[27]同样也发现未经药物治疗的抑郁患者外周血白细胞中HDAC5 mRNA水平明显上调, 经过帕罗西汀治疗8周后可恢复HDAC5 mRNA至正常水平。然而, 也有抑郁症患者尸检后结果显示伏隔核(nucleus accumbens, NAc)中H3乙酰化水平上升, HDAC2含量下降[28], 这种不一致可能与患者抑郁的状态、药物的使用以及检测样本(脑组织、血细胞)不同相关, 还有待进一步的研究确认。
另一项临床研究[29]观察到在MDD患者的外周血白细胞中SIRT (尤其是SIRT1、2和6)的表达减少, 这些改变在抑郁的缓解期恢复正常, 表明SIRT1、2和6的mRNA水平可能是情绪障碍的潜在标志物。
1.2.2 动物实验脑内奖赏系统功能障碍参与了抑郁症的发病, 该系统起源于中脑腹侧被盖区(ventral tegmental area, VTA), 通过前脑内侧束投射到NAc、前额叶皮层(prefrontal cortex, PFC)、嗅结节和杏仁核等边缘前脑区域, 组成广泛的奖赏通路。中脑-边缘多巴胺系统是奖赏系统的核心, 而NAc又是该核心的关键区。多种慢性应激已被用于动物抑郁症模型的建立, 如社交挫败应激(social defeat, SD)、慢性不可预见性应激(chronic unpredictable mild stress, CUMS)、慢性束缚应激(chronic restraint stress, CRS)和母婴分离应激等[30]。抑郁动物表现为糖水偏好下降(快感缺失)、强迫游泳实验中不动时间延长(行为绝望)以及社交回避。研究发现, 动物的抑郁样行为与某些亚型HDAC的表达改变有关, 其中针对HDAC2和HDAC5变化的研究较为集中[31]。
Erburu等[32]的研究显示, CUMS下调PFC内HDAC5和p-HDAC5的含量, Renthal等[21]观察到SD应激下调了NAc中HDAC5 mRNA的水平。SD之后发现全脑H3K14ac和H4K12ac水平上调[33]以及组蛋白H4其他几个赖氨酸位点如5、8和16的乙酰化水平的变化[34]。其中, 海马H3乙酰化的水平短暂升高, 然后持续降低[35], 同时海马中与抑郁机制密切相关的脑源性神经营养因子(brain-derived neurotrophic factor, BDNF)的mRNA水平降低, 给予抗抑郁药丙咪嗪可以增加BDNF启动子处的组蛋白乙酰化并逆转这种下降[36], 如果在海马内过表达HDAC5则会抑制丙咪嗪的抗抑郁效果。与在NAc和PFC中的作用相反, 海马中的HDAC5可能有促进抑郁症发展的作用, 因此靶向抑制海马中的HDAC5可能具有抗抑郁效果。
Nestler课题组的研究[28]发现在遭受SD后1 h, 小鼠NAc中H3乙酰化的水平暂时降低, 24 h后H3乙酰化的水平持续增加并伴随NAc中HDAC2的含量降低。与此类似, 在小鼠CRS模型中, 海马HDAC2的mRNA和蛋白水平均降低, 且给予氟西汀并不能逆转这一变化。Guan等[37]的研究显示, 神经元中过表达HDAC2可降低突触可塑性和记忆形成。据此推测NAc中HDAC2的表达减少是随着抑郁出现的一种适应性反应, 可能促进神经元可塑性, 抑制HDAC2可能成为治疗抑郁症的潜在研究方向。
这些研究表明, 疾病不同阶段可能影响HDAC的表达, HDAC及其相关机制参与了抑郁症的发展。因此, 可以推测: 调节HDAC活性, 尤其是能调节HDAC2和HDAC5活性的化合物很可能成为新型抗抑郁药物。
2 HDACi与抑郁症对HDACi的研究始于大约30年前[38], 由于HDACi可诱导细胞分化、调控细胞周期和凋亡, 并有抑制癌细胞迁移和侵袭的能力[39], 其中帕比司他(panobinostat, Farydak®)、伏立诺他(vorinostat, Zolinza®)和罗米地辛(romidepsin, Celgene®)等已作为抗癌药应用于临床[40]。
HDACi在治疗精神类疾病方面也有悠久的历史, 例如丙戊酸在发现其与HDAC的关系之前就已经在临床上主要用作情绪稳定剂[41]。也有研究证明HDACi在治疗阿尔茨海默症、帕金森等神经退行性疾病以及癫痫、精神分裂症等[42-45]精神性疾病的潜力。此外, 目前的抗抑郁药一定程度上也通过影响大脑乙酰化状态提高突触可塑性, 从而发挥抗抑郁作用[46], 因此, 开发靶向HDAC的抗抑郁药物是非常有前景的。
2.1 HDACi的分类目前的HDACi大部分都有“帽-连接基团-螯合剂”(cap-linker-chelator)的药效团结构[47], 螯合剂是指可以结合锌的基团(例如异羟肟酸酯和邻氨基苯胺); 连接基团类似赖氨酸侧链可以占据HDAC酶结合位点, 将螯合剂连接到如芳香环结构即“帽”上[48], “螯合剂”和“帽”的功能可影响酶的选择性及抑制能力[49]。HDACi可以根据结构分为以下4类[40](表 1):
| 表 1 Specific histone deacetylase inhibitors (HDACi) with potential antidepressant-activity |
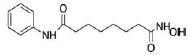

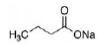
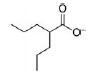

如丁酸钠(NaB)和苯基丁酸; 它们通常靶向HDAC I和HDAC II, 但其抑制HDAC活性的能力往往不如异羟肟酸类。
2.1.2 异羟肟酸类如曲古菌素A (trichostatin A, TSA)和伏立诺他(SAHA)。异羟肟酸类是数量最大的一类HDACi, 且由于对HDAC亚型的选择性低, 被称为泛HDAC抑制剂(pan-HDACi)。通常它们的结构由异羟肟酸、疏水性连接基团和大分子极性基团构成, 与HDAC的催化域结合发挥抑制作用[50]。其中, SAHA于2006年已被FDA批准用于治疗难治性或复发性的皮肤T细胞淋巴瘤(cutaneous T-cell lymphoma, CTCL)[39]。
2.1.3 环肽类如阿匹替丁(apicidine)和罗米地辛(romidepsin), 后者除了抑制HDAC I, 还激活caspase 8介导的凋亡途径[50], 被FDA批准用于外周T细胞淋巴瘤(peripheral T-cell lymphomas, PTCL)和CTCL的治疗[51]。
2.1.4 苯甲酰胺类如MS-275, 对HDAC I的选择性较强, 且由于可以选择性地改变皮层和海马的乙酰化水平而备受关注[52]。
迄今为止, 由于对特定HDAC的选择性低, 存在发生不良事件的风险, 因此, 临床上尚未将HDACi用于治疗抑郁症。本研究根据临床前研究的药效学结果, 集中讨论了几种具有最大抗抑郁潜力的HDACi (表 1[28, 35, 38-40, 50-60])。
2.2 HDACi的抗抑郁样作用和可能机制 2.2.1 短链脂肪酸NaB在小鼠CRS模型中, 外周给予NaB可以诱导海马cFOS表达并使BDNF启动子H3K9ac富集增加, 提高BDNF蛋白水平[59]。BDNF是体内含量最高的神经营养因子, 它通过与酪氨酸激酶受体(tyrosine kinase receptor B, Trk B)结合而发挥促进神经元的存活、分化和生长作用。同时NaB可以上调甲状腺素蛋白mRNA水平并下调五羟色胺2A受体(5-hydroxytryptamine 2A receptor, 5-HT2A)的mRNA水平[61], 逆转CRS诱导的海马磷酸化c AMP反应元件结合蛋白(p CREB)降低以及HDAC2、HDAC5下调[60], 这可能和其抗抑郁作用相关。此外, 对BALB/c小鼠(一种对压力易感的小鼠品系)的研究[62]表明, NaB合并氟西汀给药没有改善小鼠的焦虑状态, 但合并给药的抗抑郁作用远强于单独给予氟西汀, 未来合并HDACi给药可能成为一种有效治疗手段。
2.2.2 异羟肟酸类SAHA有文献报道, 对遭受SD的小鼠在NAc中给予SAHA显示出抗抑郁作用, 外周给予SAHA同样可以逆转CUMS应激后的抑郁样行为[53]。该作用可能是通过上调胶质细胞源性神经营养因子(glial-derived neurotrophic factor, GDNF)的表达从而改善抑郁。Kv等[54]发现预防性连续14天腹腔注射SAHA, 可降低慢性皮质酮(corticosterone, CORT)诱发的应激模型小鼠血清中皮质酮的含量, 减轻其焦虑和行为绝望状态, 并且SAHA治疗能够阻断CORT诱导的海马中TNF-α和IL-1β水平升高, 这可能是由于SAHA干扰了转录因子募集的作用, 从而抑制了Toll样受体(Toll-like receptor, TLR)诱导细胞因子和趋化因子的过程, 并作用于炎症相关的NF-κB和JNK通路[55]。目前, 已有大量的研究显示了抑郁症与炎症的密切关系: 抑郁症患者的促炎细胞因子、C反应蛋白、趋化因子和细胞黏附分子水平较高[63], 且使用干扰素-α治疗后, 患者抑郁症的发生率高达50%[64], 因此SAHA可能是通过减轻炎症性损伤从而改善抑郁症状。此外, SAHA还显示出治疗难治性抑郁症的作用: 小鼠缺少CREB调控的转录共激活因子1 (CRTC1), 其抑郁症状不能为氟西汀所逆转[65], 但外周给予SAHA可以部分逆转Crtc1-/-小鼠的抑郁样行为, 同时提高海马以及PFC中H3、H4乙酰化水平, 并伴随着这些小鼠PFC中BDNF的表达增加[56]。
2.2.3 苯甲酰胺类MS-275Nestler团队的研究[28]显示, 在NAc中连续5天给予MS-275可以逆转SD引起的社交回避、糖水偏爱下降、强迫游泳中不动时间延长的行为。微阵列(microarray)测序结果显示, SD诱导的NAc中特殊基因表达模式经氟西汀治疗后被逆转, NAc中连续给予10天MS-275也有类似于氟西汀的逆转作用。但两者调控的基因并不完全重合, 提示被MS-275选择性调节的基因可能为抗抑郁疗法提供有希望的新型靶标。
类似地, 如果将MS-275注入海马可逆转SD引起的糖水偏爱下降, 但并不能恢复社交互动行为, 有趣的是, 如果将MS-275直接注入杏仁核或PFC后, 社交回避现象会得到逆转, 但不能改变糖水偏爱下降[35], 提示不同脑区组蛋白乙酰化可能只参与了部分抑郁样行为的调节, 这对研究者了解不同脑区在抑郁发病过程中的作用具有重要意义。
2.2.4 SIRT抑制剂有临床研究表明, 抑郁症患者外周血中SIRT1的表达明显低于健康受试者[66]。AbeHiguchi等[67]发现, 持续给予6周的慢性超轻度应激(chronic ultra-mild stress)降低了小鼠海马齿状回(den‐tate gyrus, DG)中SIRT1的蛋白水平和活性, SIRT1抑制剂sirtinol持续14天注入正常BALB/c小鼠的DG会诱发其产生抑郁样行为。然而Ferland等[68]的研究发现, CUMS造成大鼠抑郁状态的同时, 上调了DG中的SIRT1活性、降低了组蛋白乙酰化水平, 向大鼠DG预防性注入sirtinol 10天后, 可以阻断上述行为学和分子的变化。作者认为这种不一致可能是因为SIRT表达具有昼夜节律性, 而慢性应激可能改变其节律; 此外, 采集组织的时间不同也可能是原因之一。因此, 海马中的SIRT1通路介导的与抑郁相关的生理过程还有待进一步研究。
Erburu等[69]发现选择性的SIRT2抑制剂33i逆转了CUMS引起的快感缺失和社交回避现象。后续研究证明, 33i上调PFC中NMDA受体亚基Glu N2A和Glu N2B的表达, 提示33i促进了谷氨酸突触传递, 文章指出该作用通过PFC中谷氨酸和5-HT系统的调节来介导, 提示SIRT2抑制剂的抗抑郁潜力。
2.2.5 HDAC6抑制剂ACY-738氟西汀等传统抗抑郁药起效慢(大部分患者需要至少2周才开始起效)一直是临床使用上的一大问题[70], 然而实验显示外周给予ACY-738 0.5 h就能够显著减少抑郁小鼠在强迫游泳测试中的不动时间, 合并给予行为无效剂量的ACY-738与亚治疗剂量的西酞普兰所产生的药效, 相当于单独给予40倍治疗剂量西酞普兰所产生的药效。HDAC6的抗抑郁作用可能部分是通过解除对5-HT介导信号传导的抑制作用来实现的[71]。此外, 抑制HDAC6可以调节糖皮质激素受体的核运输, 并减少中缝背核内5-HT能神经元和PFC内锥体神经元的应激信号传导, 从而促进突触可塑性[72], 因此, 抑制HDAC6也是一种有前景的快速抗抑郁策略。
2.2.6 新型HDAC I抑制剂IN14IN14是一种可以透过血脑屏障的新型HDAC I抑制剂, 且盐沼曲霉测试显示其低毒性特质。强迫游泳测试显示: 给予5天的IN14, 比三环类抗抑郁药地昔帕明具有更好的抗抑郁作用, 此外, IN14能诱导小鼠产生更多的攀爬动作, 表明其具有促动机行为作用, IN14可能代表了一种有前途的新型低毒抗抑郁药[73]。
3 总结与展望综上, HDACi可能通过3种潜在机制发挥抗抑郁的作用: ① HDACi可以调节炎症相关的NF-κB和JNK通路, 减轻炎症损伤和氧化应激; ②通过调节谷氨酸和5-HT系统改善谷氨酸传递; ③通过上调神经保护性或神经营养性蛋白或诸如GDNF和BDNF等因子的表达, 起到神经保护作用(图 1)。

|
Figure 1 The potential mechanism of antidepressant-activity of histone deacetylase inhibitors (HDACi).Chromatin can be conceptualized as existing in two primary structural states: as active euchromatin in which histone acetylation is associated with opening the nucleosome to allow binding of the basal transcriptional complex, or as inactive heterochromatin where all gene activity is permanently silenced.Transcrip‐tional dysregulation can be induced by stress and leads hypoacetylation status which decrease the transcription of brain-derived neurotrophic factor (BDNF) and other neuronal growth factors involved in the development of depression.While HDACi play the role in anti-depression by inhibiting the activity of HDACs, shifting the balance toward active transcription of the set of interrelated genes |
然而, 目前HDACi在临床上的使用仍然存在争议, 这些化合物通常由于特异性低且透过血脑屏障的能力有限, 往往需要较高剂量才能起作用。针对HDACi特异性较低的问题, 目前能克服此问题的化合物之一就是MS-275, 它可以选择性阻断HDAC I的活性且可以透过血脑屏障。另一方面, 有研究表明即使在低剂量使用情况下, MS-275、NaB或ACY-738也可以增强经典抗抑郁药(特别是氟西汀)的功效[74]。且HDACi往往可在7天之内发挥药效, 而氟西汀或其他标准抗抑郁药(例如丙咪嗪)则需要14~20天才能观察到抗抑郁效果[75]。因此, 针对HDACi的研究不仅提高了研究者对HDAC在抑郁症中所发挥作用的认识, 还为理解抑郁和抗抑郁作用的潜在分子机制提供了新的思路, 并支持了HDACi可能在染色质结构水平起作用的抗抑郁潜力, 值得在临床中进行下一步研究。
表观遗传学机制在一定程度上拓展了抑郁症病因学研究视野, 提供了一种解释环境因素影响基因表达的可能机制。HDAC可能代表了研究人员一直在寻找的解释抑郁症等疾病发病机制的缺失环节, 填补了遗传学无法解释的空白。未来, 研究人员仍需要阐明HDACi功能的确切分子机制, 确定是否有可能靶向大脑中的特定HDAC亚型, 以便探索更加有效的新治疗策略。
作者贡献:姚开云进行了全文的撰写以及图片、表格的制作; 丁虹琬、曹琳玉、高银阁和张建军参与文章校对与修改; 本文的通讯作者为王贵彬, 提供文章思路, 负责主要的审阅与修改工作。
利益冲突:无利益冲突。
| [1] |
Smith K. Mental health: a world of depression[J]. Nature, 2014, 515: 181. |
| [2] |
Naqvi TZ, Naqvi SS, Merz CN. Gender differences in the link between depression and cardiovascular disease[J]. Psychosom Med, 2005, 67 Suppl 1: S15-S18. |
| [3] |
Berton O, Nestler EJ. New approaches to antidepressant drug discovery: beyond monoamines[J]. Nat Rev Neurosci, 2006, 7: 137-151. DOI:10.1038/nrn1846 |
| [4] |
Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longerterm outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report[J]. Am J Psychiatry, 2006, 163: 1905-1917. DOI:10.1176/ajp.2006.163.11.1905 |
| [5] |
Molero P, Ramos-Quiroga JA, Martin-Santos R, et al. Antidepressant efficacy and tolerability of ketamine and esketamine: a critical review[J]. CNS Drugs, 2018, 32: 411-420. DOI:10.1007/s40263-018-0519-3 |
| [6] |
Power RA, Tansey KE, Buttenschon HN, et al. Genome-wide association for major depression through age at onset stratification: major depressive disorder working group of the psychiatric genomics consortium[J]. Biol Psychiatry, 2017, 81: 325-335. DOI:10.1016/j.biopsych.2016.05.010 |
| [7] |
Howard DM, Adams MJ, Clarke TK, et al. Genome-wide metaanalysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions[J]. Nat Neurosci, 2019, 22: 343-352. DOI:10.1038/s41593-018-0326-7 |
| [8] |
Berger SL, Kouzarides T, Shiekhattar R, et al. An operational definition of epigenetics[J]. Genes Dev, 2009, 23: 781-783. DOI:10.1101/gad.1787609 |
| [9] |
Stein MB, Jang KL, Taylor S, et al. Genetic and environmental influences on trauma exposure and posttraumatic stress disorder symptoms: a twin study[J]. Am J Psychiatry, 2002, 159: 1675-1681. DOI:10.1176/appi.ajp.159.10.1675 |
| [10] |
Nestler EJ. Epigenetic mechanisms of depression[J]. JAMA Psychiatry, 2014, 71: 454-456. DOI:10.1001/jamapsychiatry.2013.4291 |
| [11] |
Gold PW. The organization of the stress system and its dysregulation in depressive illness[J]. Mol Psychiatry, 2015, 20: 32-47. DOI:10.1038/mp.2014.163 |
| [12] |
Bagot RC, Labonte B, Pena CJ, et al. Epigenetic signaling in psychiatric disorders: stress and depression[J]. Dialogues Clin Neurosci, 2014, 16: 281-295. DOI:10.31887/DCNS.2014.16.3/rbagot |
| [13] |
Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy[J]. Nat Rev Genet, 2009, 10: 32-42. DOI:10.1038/nrg2485 |
| [14] |
Clayton AL, Hazzalin CA, Mahadevan LC. Enhanced histone acetylation and transcription: a dynamic perspective[J]. Mol Cell, 2006, 23: 289-296. DOI:10.1016/j.molcel.2006.06.017 |
| [15] |
Grunstein M. Histone acetylation in chromatin structure and transcription[J]. Nature, 1997, 389: 349-352. DOI:10.1038/38664 |
| [16] |
Sabari BR, Zhang D, Allis CD, et al. Metabolic regulation of gene expression through histone acylations[J]. Nat Rev Mol Cell Biol, 2017, 18: 90-101. DOI:10.1038/nrm.2016.140 |
| [17] |
Shen Y, Wei W, Zhou DX. Histone acetylation enzymes coordinate metabolism and gene expression[J]. Trends Plant Sci, 2015, 20: 614-621. DOI:10.1016/j.tplants.2015.07.005 |
| [18] |
Yang YW, Shuang S, Hu ZW, et al. Advances and applications of lysine acetylation[J]. Acta Pharm Sin(药学学报), 2019, 54: 778-787. |
| [19] |
Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men[J]. Nat Rev Mol Cell Biol, 2008, 9: 206-218. |
| [20] |
Porter NJ, Christianson DW. Structure, mechanism, and inhibition of the zinc-dependent histone deacetylases[J]. Curr Opin Struct Biol, 2019, 59: 9-18. DOI:10.1016/j.sbi.2019.01.004 |
| [21] |
Renthal W, Maze I, Krishnan V, et al. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli[J]. Neuron, 2007, 56: 517-529. DOI:10.1016/j.neuron.2007.09.032 |
| [22] |
Gore SD, Carducci MA. Modifying histones to tame cancer: clinical development of sodium phenylbutyrate and other histone deacetylase inhibitors[J]. Expert Opin Investig Drugs, 2000, 9: 2923-2934. DOI:10.1517/13543784.9.12.2923 |
| [23] |
Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes[J]. Cold Spring Harb Perspect Biol, 2014, 6: a018713. DOI:10.1101/cshperspect.a018713 |
| [24] |
Schwer B, Verdin E. Conserved metabolic regulatory functions of sirtuins[J]. Cell Metab, 2008, 7: 104-112. DOI:10.1016/j.cmet.2007.11.006 |
| [25] |
Liu H, Hu Q, Kaufman A, et al. Developmental expression of histone deacetylase 11 in the murine brain[J]. J Neurosci Res, 2008, 86: 537-543. DOI:10.1002/jnr.21521 |
| [26] |
Hobara T, Uchida S, Otsuki K, et al. Altered gene expression of histone deacetylases in mood disorder patients[J]. J Psychiatr Res, 2010, 44: 263-270. DOI:10.1016/j.jpsychires.2009.08.015 |
| [27] |
Iga J, Ueno S, Yamauchi K, et al. Altered HDAC5 and CREB mRNA expressions in the peripheral leukocytes of major depression[J]. Prog Neuropsychopharmacol Biol Psychiatry, 2007, 31: 628-632. DOI:10.1016/j.pnpbp.2006.12.014 |
| [28] |
Covington HE 3rd, Maze I, LaPlant QC, et al. Antidepressant actions of histone deacetylase inhibitors[J]. J Neurosci, 2009, 29: 11451-11460. DOI:10.1523/JNEUROSCI.1758-09.2009 |
| [29] |
Abe N, Uchida S, Otsuki K, et al. Altered sirtuin deacetylase gene expression in patients with a mood disorder[J]. J Psychiatr Res, 2011, 45: 1106-1112. DOI:10.1016/j.jpsychires.2011.01.016 |
| [30] |
Gururajan A, Reif A, Cryan JF, et al. The future of rodent models in depression research[J]. Nat Rev Neurosci, 2019, 20: 686-701. DOI:10.1038/s41583-019-0221-6 |
| [31] |
Penney J, Tsai LH. Histone deacetylases in memory and cognition[J]. Sci Signal, 2014, 7: re12. DOI:10.1126/scisignal.aaa0069 |
| [32] |
Erburu M, Cajaleon L, Guruceaga E, et al. Chronic mild stress and imipramine treatment elicit opposite changes in behavior and in gene expression in the mouse prefrontal cortex[J]. Pharmacol Biochem Behav, 2015, 135: 227-236. DOI:10.1016/j.pbb.2015.06.001 |
| [33] |
Chou AH, Chen YL, Hu SH, et al. Polyglutamine-expanded ataxin-3 impairs long-term depression in Purkinje neurons of SCA3 transgenic mouse by inhibiting HAT and impairing histone acetylation[J]. Brain Res, 2014, 1583: 220-229. DOI:10.1016/j.brainres.2014.08.019 |
| [34] |
Kenworthy CA, Sengupta A, Luz SM, et al. Social defeat induces changes in histone acetylation and expression of histone modifying enzymes in the ventral hippocampus, prefrontal cortex, and dorsal raphe nucleus[J]. Neuroscience, 2014, 264: 88-98. DOI:10.1016/j.neuroscience.2013.01.024 |
| [35] |
Covington HE 3rd, Vialou VF, LaPlant Q, et al. Hippocampaldependent antidepressant-like activity of histone deacetylase inhibition[J]. Neurosci Lett, 2011, 493: 122-126. DOI:10.1016/j.neulet.2011.02.022 |
| [36] |
Tsankova NM, Berton O, Renthal W, et al. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action[J]. Nat Neurosci, 2006, 9: 519-525. DOI:10.1038/nn1659 |
| [37] |
Guan JS, Haggarty SJ, Giacometti E, et al. HDAC2 negatively regulates memory formation and synaptic plasticity[J]. Nature, 2009, 459: 55-60. DOI:10.1038/nature07925 |
| [38] |
Szyf M. Prospects for the development of epigenetic drugs for CNS conditions[J]. Nat Rev Drug Discov, 2015, 14: 461-474. DOI:10.1038/nrd4580 |
| [39] |
McClure JJ, Li X, Chou CJ. Advances and challenges of HDAC inhibitors in cancer therapeutics[J]. Adv Cancer Res, 2018, 138: 183-211. |
| [40] |
Ververis K, Hiong A, Karagiannis TC, et al. Histone deacetylase inhibitors(HDACIs): multitargeted anticancer agents[J]. Biologics, 2013, 7: 47-60. |
| [41] |
Haddad PM, Das A, Ashfaq M, et al. A review of valproate in psychiatric practice[J]. Expert Opin Drug Metab Toxicol, 2009, 5: 539-551. DOI:10.1517/17425250902911455 |
| [42] |
Kelly DL, Conley RR, Feldman S, et al. Adjunct divalproex or lithium to clozapine in treatment-resistant schizophrenia[J]. Psychiatr Q, 2006, 77: 81-95. DOI:10.1007/s11126-006-7963-9 |
| [43] |
Fischer A, Sananbenesi F, Mungenast A, et al. Targeting the correct HDAC(s) to treat cognitive disorders[J]. Trends Pharmacol Sci, 2010, 31: 605-617. DOI:10.1016/j.tips.2010.09.003 |
| [44] |
Suo H, Wang P, Tong J, et al. NRSF is an essential mediator for the neuroprotection of trichostatin A in the MPTP mouse model of Parkinson's disease[J]. Neuropharmacology, 2015, 99: 67-78. DOI:10.1016/j.neuropharm.2015.07.015 |
| [45] |
Citraro R, Leo A, De Caro C, et al. Effects of histone deacetylase inhibitors on the development of epilepsy and psychiatric comorbidity in WAG/Rij rats[J]. Mol Neurobiol, 2020, 57: 408-421. DOI:10.1007/s12035-019-01712-8 |
| [46] |
Erburu M, Munoz-Cobo I, Dominguez-Andres J, et al. Chronic stress and antidepressant induced changes in HDAC5 and SIRT2affect synaptic plasticity[J]. Eur Neuropsychopharmacol, 2015, 25: 2036-2048. DOI:10.1016/j.euroneuro.2015.08.016 |
| [47] |
Bowers AA, Greshock TJ, West N, et al. Synthesis and conformation-activity relationships of the peptide isosteres of FK228 and largazole[J]. J Am Chem Soc, 2009, 131: 2900-2905. DOI:10.1021/ja807772w |
| [48] |
Bradner JE, West N, Grachan ML, et al. Chemical phylogenetics of histone deacetylases[J]. Nat Chem Biol, 2010, 6: 238-243. DOI:10.1038/nchembio.313 |
| [49] |
Arrowsmith CH, Bountra C, Fish PV, et al. Epigenetic protein families: a new frontier for drug discovery[J]. Nat Rev Drug Discov, 2012, 11: 384-400. DOI:10.1038/nrd3674 |
| [50] |
Grayson DR, Kundakovic M, Sharma RP. Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders?[J]. Mol Pharmacol, 2010, 77: 126-135. DOI:10.1124/mol.109.061333 |
| [51] |
Frye R, Myers M, Axelrod KC, et al. Romidepsin: a new drug for the treatment of cutaneous T-cell lymphoma[J]. Clin J Oncol Nurs, 2012, 16: 195-204. DOI:10.1188/12.CJON.195-204 |
| [52] |
Simonini MV, Camargo LM, Dong E, et al. The benzamide MS-275 is a potent, long-lasting brain region-selective inhibitor of histone deacetylases[J]. Proc Natl Acad Sci U S A, 2006, 103: 1587-1592. DOI:10.1073/pnas.0510341103 |
| [53] |
Uchida S, Hara K, Kobayashi A, et al. Epigenetic status of GDNF in the ventral striatum determines susceptibility and adaptation to daily stressful events[J]. Neuron, 2011, 69: 359-372. DOI:10.1016/j.neuron.2010.12.023 |
| [54] |
Kv A, Madhana RM, Js IC, et al. Antidepressant activity of vorinostat is associated with amelioration of oxidative stress and inflammation in a corticosterone-induced chronic stress model in mice[J]. Behav Brain Res, 2018, 344: 73-84. DOI:10.1016/j.bbr.2018.02.009 |
| [55] |
Bode KA, Schroder K, Hume DA, et al. Histone deacetylase inhibitors decrease Toll-like receptor-mediated activation of proinflammatory gene expression by impairing transcription factor recruitment[J]. Immunology, 2007, 122: 596-606. DOI:10.1111/j.1365-2567.2007.02678.x |
| [56] |
Meylan EM, Halfon O, Magistretti PJ, et al. The HDAC inhibitor SAHA improves depressive-like behavior of CRTC1-deficient mice: possible relevance for treatment-resistant depression[J]. Neuropharmacology, 2016, 107: 111-121. DOI:10.1016/j.neuropharm.2016.03.012 |
| [57] |
Chen WY, Zhang H, Gatta E, et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) alleviates depression-like behavior and normalizes epigenetic changes in the hippocampus during ethanol withdrawal[J]. Alcohol, 2019, 78: 79-87. DOI:10.1016/j.alcohol.2019.02.005 |
| [58] |
Valvassori SS, Resende WR, Budni J, et al. Sodium butyrate, a histone deacetylase inhibitor, reverses behavioral and mitochondrial alterations in animal models of depression induced by earlyor late-life stress[J]. Curr Neurovasc Res, 2015, 12: 312-320. DOI:10.2174/1567202612666150728121121 |
| [59] |
Schroeder FA, Lin CL, Crusio WE, et al. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse[J]. Biol Psychiatry, 2007, 62: 55-64. DOI:10.1016/j.biopsych.2006.06.036 |
| [60] |
Han A, Sung YB, Chung SY, et al. Possible additional antidepressant-like mechanism of sodium butyrate: targeting the hippocampus[J]. Neuropharmacology, 2014, 81: 292-302. DOI:10.1016/j.neuropharm.2014.02.017 |
| [61] |
Yamawaki Y, Fuchikami M, Morinobu S, et al. Antidepressantlike effect of sodium butyrate (HDAC inhibitor) and its molecular mechanism of action in the rat hippocampus[J]. World J Biol Psychiatry, 2012, 13: 458-467. DOI:10.3109/15622975.2011.585663 |
| [62] |
Schmauss C. An HDAC-dependent epigenetic mechanism that enhances the efficacy of the antidepressant drug fluoxetine[J]. Sci Rep, 2015, 5: 8171. DOI:10.1038/srep08171 |
| [63] |
Leighton SP, Nerurkar L, Krishnadas R, et al. Chemokines in depression in health and in inflammatory illness: a systematic review and meta-analysis[J]. Mol Psychiatry, 2018, 23: 48-58. DOI:10.1038/mp.2017.205 |
| [64] |
Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression[J]. Trends Immunol, 2006, 27: 24-31. DOI:10.1016/j.it.2005.11.006 |
| [65] |
Breuillaud L, Rossetti C, Meylan EM, et al. Deletion of CREBregulated transcription coactivator 1 induces pathological aggression, depression-related behaviors, and neuroplasticity genes dysregulation in mice[J]. Biol Psychiatry, 2012, 72: 528-536. DOI:10.1016/j.biopsych.2012.04.011 |
| [66] |
Lu G, Li J, Zhang H, et al. Role and possible mechanisms of Sirt1 in depression[J]. Oxid Med Cell Longev, 2018, 2018: 8596903. |
| [67] |
Abe-Higuchi N, Uchida S, Yamagata H, et al. Hippocampal sirtuin 1 signaling mediates depression-like behavior[J]. Biol Psychiatry, 2016, 80: 815-826. DOI:10.1016/j.biopsych.2016.01.009 |
| [68] |
Ferland CL, Hawley WR, Puckett RE, et al. Sirtuin activity in dentate gyrus contributes to chronic stress-induced behavior and extracellular signal-regulated protein kinases 1 and 2 cascade changes in the hippocampus[J]. Biol Psychiatry, 2013, 74: 927-935. DOI:10.1016/j.biopsych.2013.07.029 |
| [69] |
Erburu M, Munoz-Cobo I, Diaz-Perdigon T, et al. SIRT2 inhibition modulate glutamate and serotonin systems in the prefrontal cortex and induces antidepressant-like action[J]. Neuropharmacology, 2017, 117: 195-208. DOI:10.1016/j.neuropharm.2017.01.033 |
| [70] |
Nierenberg AA, Farabaugh AH, Alpert JE, et al. Timing of onset of antidepressant response with fluoxetine treatment[J]. Am J Psychiatry, 2000, 157: 1423-1428. DOI:10.1176/appi.ajp.157.9.1423 |
| [71] |
Fukada M, Hanai A, Nakayama A, et al. Loss of deacetylation activity of HDAC6 affects emotional behavior in mice[J]. PLoS One, 2012, 7: e30924. DOI:10.1371/journal.pone.0030924 |
| [72] |
Espallergues J, Teegarden SL, Veerakumar A, et al. HDAC6 regulates glucocorticoid receptor signaling in serotonin pathways with critical impact on stress resilience[J]. J Neurosci, 2012, 32: 4400-4416. DOI:10.1523/JNEUROSCI.5634-11.2012 |
| [73] |
Martinez-Pacheco H, Picazo O, Lopez-Torres A, et al. Biochemical and behavioral characterization of IN14, a new inhibitor of HDACs with antidepressant-like properties[J]. Biomolecules, 2020, 10: 299. DOI:10.3390/biom10020299 |
| [74] |
Ghosh B, Zhao WN, Reis SA, et al. Dissecting structure-activityrelationships of crebinostat: brain penetrant HDAC inhibitors for neuroepigenetic regulation[J]. Bioorg Med Chem Lett, 2016, 26: 1265-1271. DOI:10.1016/j.bmcl.2016.01.022 |
| [75] |
Covington HE 3rd, Maze I, Vialou V, et al. Antidepressant action of HDAC inhibition in the prefrontal cortex[J]. Neuroscience, 2015, 298: 329-335. DOI:10.1016/j.neuroscience.2015.04.030 |