 2020, Vol. 55
2020, Vol. 55

新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
本刊在2014年刊载了孟鲁司特的研发历程, 与本条目扎鲁司特都是白三烯受体拮抗剂, 口服治疗哮喘病的药物。本品上市于1996年, 早于孟鲁司特两年, 但都具有首创性特征, 在没有受体结构的参考下, 各自凭借药物化学的试错方法(trial and error)和构效关系(SAR), 由简单的苗头分子演绎并优化成功。其实, 扎鲁司特的苗头分子就有白三烯的结构痕迹, 构成与受体结合的基础, 并且在结构优化中始终保持分子中的亲脂链和酸性基团, 以满足受体对药效团的要求, 所以对于受体结构未知的新药创制, 本案例具有参考价值。 (编者按)
上世纪30年代发现动物致敏后体内产生不稳定的化学物质, 称作慢反应物质, 1960年Brockelhurst揭示这是免疫系统在受到抗原攻击产生的内源性物质, 可引起平滑肌(如气道和十二指肠)收缩, 称之为过敏性慢反应物质(SRS-A) (Brocklehurst WE. The release of histamine and formation of a slow reacting substance (SRS-A) during anaphylactic shock. J Physiol, 1960, 151: 416-435)。20世纪70年代证实SRS-A是由花生四烯酸(AA)代谢生成的3个肽白三烯(peptidyl leucotrienes) (简称白三烯), 即LTC4、LTD4和LTE4, 三者之差异是在二十碳四烯酸中分别连接3、2、1个氨基酸(图 1)。白三烯的生理作用是引起呼吸道平滑肌收缩和增加肺泡的通透性以及黏膜上皮细胞分泌等, 导致哮喘和过敏性疾病。

|
Figure 1 Structure of peptide leukotriene |
20世纪80年代ICI公司(今AstraZeneca)启动白三烯受体拮抗剂项目时, 只有以Fisons公司研制的FPL55712 (1)为代表的化合物(Appleton RA, Bantick JR, Chamberlain TR, et al. Antagonists of slow reacting substance of anaphylaxis. Synthesis of a series of chromone-2-carboxylic acids. J Med Chem, 1977, 20: 371-379), 1的结构特征是含羧基的极性端, 另一端为疏水性基团, 映射了受体与激动剂结合的结构特征。

|
用豚鼠支气管条作为实验材料, 在生理浴液中将支气管条固定在张力测定仪上, 加入10 nmol·L-1激动剂LTE4 (或LTD4)引起收缩, 达到平衡后记录张力。加入一定浓度的受试物, 平衡后记录张力的变化, 与空白对照, 计算化合物抑制LTE4收缩作用的百分抑制率。
2.2 体外离解常数对上述舒张活性较强的化合物用豚鼠支气管条测定离解常数KB, 记录不同浓度的受试物舒张LTE4的收缩作用, 计算降低最大收缩作用的一半(以30 μmol·L-1氨甲酰胆碱引起的收缩力为100%)的受试物浓度为KB值。
2.3 体内实验将豚鼠头部密闭于含有一定浓度LTD4的气雾装置中, 引起呼吸短促与困难。在此之前给受试物, 测定的终末点是达到平稳得以自由的慢速呼吸时所需的时间, 与空白组的时间对比计算受试物的保护作用(百分数)。
3 先导化合物的产生 3.1 从白三烯结构入手图 1中白三烯的结构可划分成三部分: C1~C6的羧酸极性部分、含有4个烯键的亲脂部分和氨基酸部分。研发者模拟前两部分的结构特征, 设计合成了一系列苄醚化合物, 其中化合物2显示弱活性, 在5 μmol·L-1浓度下抑制活性达40%, pKB 5.1。

|
亲脂性的正己氧基与羧基的空间位置对提高活性很重要, 但苄醚的柔性键不利于与受体结合, 因而将连接两个苯环的片段环合以固定片段间的空间位置, 在并合环的不同位置连接亲脂链和羧基, 以调整相对位置, 在“固化”的构象中优化活性。以吲唑为并合的母核, N1和N2可作为调节苯甲酸空间位置的连接位点。由于硝基吲唑是易得原料, 从而演化成酰胺作为连接基引出亲脂链, 如通式3所示。依此通式合成的化合物列于表 1。
| Table 1 Structure and activity of heptanoyl amidoindazole-benzyl acids (10 μmol·L-1) |
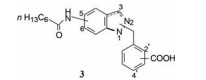
表 1的8个化合物互为区域异构体, 其中只有化合物9显示活性, 羧基在苯环的对位, 不同于羧基在邻位的化合物2。苄基连接在N2的化合物10和11没有活性, 提示N2没有必要作为连接位点, 可用吲哚替换。
3.3 吲哚替换吲唑环另一方面, 参照色酮类(化合物1)的构效关系, 当苄基上羧基的间位存在甲氧基取代, 比未取代的活性显著提高, 推论9的2'位引入甲氧基也可能有增效作用, 从而合成了吲哚为母核的化合物12, 实验表明活性显著提高, 浓度为5 μmol·L-1的抑制率达到70%, 而且对白三烯受体的选择性作用也提高了。12作为先导化合物进行优化。
4 吲哚化合物的优化 4.1 酰基的变换化合物12的N1-2'-甲氧基-4'-羧苄基不变, 变换6位的酰基, 构效关系表明正丁酰基(13, 5 μmol·L-1的抑制率42%)和正庚酰基的活性化合物12相近; 苯甲酰基和苯烷酰基化合物中只有苯乙酰化合物14活性与12相当(5 μmol·L-1的抑制率71%)。

|
化合物12~14是链烷酰和芳烷酰基的优选分子, 在此基础上考察链的支化对活性的影响。合成的化合物列于表 2。
| Table 2 Activities of compounds with branched acyl groups |

化合物15的设计意图是侧链包含了13的正丁酰和14的苯乙酰片段, 实验结果表明15的活性提高了5倍, 推论13和14的侧链结合于不同的疏水腔, 由于15的侧链支化, 分别结合于不同的脂性腔穴中(需进一步证明), 加和作用提高了活性。然而其他的支化链化合物16~20都降低了活性。21~24也都没有超过12的活性。所以, 酰基侧链的α位取代无助于提高活性。S构型的24活性强于R构型, 提示该侧链的结合具有立体选择性。
4.3 酰胺的变换侧链的-CONH-连接基用N-烷基、碱性胺基、硫代酰胺基、脲基或胺甲酰基等置换, 化合物及其活性列于表 3。
| Table 3 Activities of compounds with different linkers at the side chain |
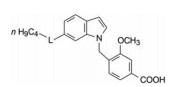
表 3中连接基为脲或胺甲酸酯的化合物28和29, 活性比12提高6倍, 提示了新的连接基可作为优化基团。然而以脲为连接基, 变换的烷基只有环戊基(30, 70%/1 μmol·L-1)显示较高活性。
4.4 氨基甲酸酯为连接基用氨基甲酸酯作连接基合成的化合物列于表 4。构效关系提示, 尺寸小的亲脂基团或直链烷基活性都比较低, 而异丙基和3-戊基显著提高活性, 提示支化有利于活性, 环丁基(37)特别是环戊基(36)的活性更高, 而环上嵌入氧原子(39)不利于活性。
| Table 4 Activities of compounds with varied urethane ester groups |
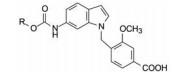
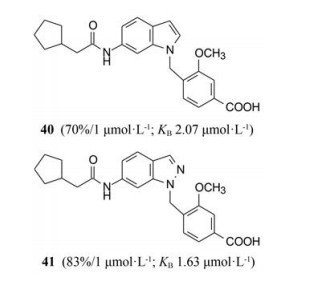
化合物30和36都由于含环戊基而活性显著高于同系列其他基团, 因而制备了环戊乙酰胺化合物40和以吲唑为母核的化合物41, 它们确实都显示有活性(Brown FJ, Yee YK, Cronk LA, et al. Evolution of a series of peptidoleukotriene antagonists: synthesis and structure-activity relationships of 1, 6-disubstituted indoles and indazoles. J Med Chem, 1990, 33: 1771-1881)。

|
|
优化至此, 以吲哚(或吲唑)为母核的优选结构是① N1连接的2'-甲氧基-4'羧基甲苯; ② 6位连接环戊乙酰胺或有支链的脂肪酰胺。其实, 将苄基从N1移至3位, 药效团的空间配置并没有显著区别, 但电性分布则有变化, 例如分子的偶极矩和静电势的改变, 从而有可能改变结合状态。这是在受体结构未知情况下常用的药物化学方法。
5.1 N1取代基的变换将化合物12的2'-甲氧基-4'羧基甲苯移至吲哚母核的3位, 化合物42的活性与12 (70%/5 μmol·L-1)相近, N1被亲脂性基团取代, 活性提高, 尤其是甲基化的43更为显著, 但N1连接极性基团(46)则失去活性(表 5)。由于N1-甲基化有利于活性, 故以后优化其他部位时, 都固定为N1-CH3。
| Table 5 The activity of the compound that changes the N1 substituent |
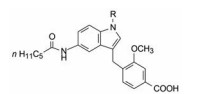
基于前述的构效关系, 酰基只在环戊基和有支链的烷基范围内变换, 表 6列出的数据提示, 以环戊基为侧链的化合物活性显著高于前面述及的化合物系列。化合物48的Ki值高达80 nmol·L-1, 而相应脲基化合物49的活性弱10倍。
| Table 6 Activity of compounds that convert acyl groups. *Inhibition constant is determined by displacement experiment of guinea pig bronchial smooth muscle cell membrane for [3H]LTD4 binding |
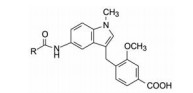
分子结构中某部分的变换, 由于形状或电性的改变会使另一部分业已优化的结合发生改变, 这是药物构效关系经常发生的现象。下面的优化是侧链为环戊乙酰基或氨基甲酸环戊醇酯时, 考察N1取代基对活性的影响。
表 7列出的构效关系表明, 氨基甲酸酯化合物47的N-CH3被H置换(50), 虽然极性加大, 但仍保持活性; 加大亲脂性基团仍有较高活性。同样酰胺系列的55~59当N1被亲脂性基团取代也有很高的活性, 尤其是59的N-苄基化合物活性提高了10倍。
| Table 7 Activities of compounds with varied both N1 substituents and C5 side chains |
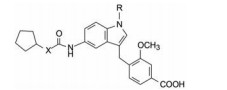
化学上羧基有许多可置换的酸性等排体, 酰胺若再被磺酰化(需有N-H), 氮上氢原子同时被酰基和磺酰基拉电子, 成为酸性氢, pKa接近于羧基。因而合成了苯磺酰胺化合物, 表 8列出了化合物的结构与活性。
| Table 8 Activities of benzenesulfonamide compounds. *Gavage to guinea pigs |
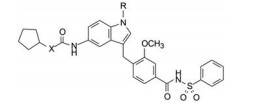
表 8的数据表明, 羧基变换为磺酰胺活性显著提高, 例如化合物61比相应的47活性提高了100倍。但这与N1的取代基有关, 例如N-正丙基63与51的活性只相差5倍, N-环戊基的64与54的活性差别就很小。这个现象未必是N1基团的位阻所致, 因为化合物62的N-CH2CH2OCH3的体积也不小, 活性却很高, 或许是极性氧原子有利地调整了疏水性。化合物66是以脲为连接基的磺酰胺化合物, 活性比相应的羧基化合物(Ki = 1 119 nmol·L-1)高1 000倍, 苯磺酰胺酰基替换了羧基使66的结合能增加了4.2 kcal·mol-1。
综上, 化合物61、62、63和65都有较高的体外活性, 体内灌胃给药, 对LTD4喷雾造成呼吸困难的豚鼠, 在低剂量下能有效地阻断LTD4诱导的呼吸困难(表 8)。
6.2 苯磺酰基的环上取代为了优化苯磺酰基的结构, 在苯基的2位引入甲基、氯和溴, 考察体外和体内的活性。表 9列出了体内外活性, 结果表明在苯环的邻位引入甲基或卤素, 体外活性没有明显改变, 但提高了体内对抗LTD4的活性。例如化合物68的半数有效剂量比没有甲基的相应化合物61提高了10倍。
| Table 9 The effect of substituents at benzenesulfonyl ring on activities |
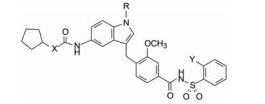
化合物68有很高的体外活性(Ki = 0.3 nmol·L-1), 体内的半数有效剂量ED50 = 0.2 mg·kg-1, 溶解性也优于其他化合物, 豚鼠灌胃吸收后血浆消除半衰期t1/2 > 14 h。选择性方面, 10 μmol·L-1浓度下68对多种膜蛋白受体(肾上腺能α、β、H1、H2、5-HT2、PGE2等)没有显示活性。因而确定为候选化合物, 定名为扎鲁司特(zafirlukast), 经临床前和临床研究表明是治疗和预防哮喘的口服用药。于1996年美国FDA批准上市。
|