 2020, Vol. 55
2020, Vol. 55

囊性纤维化是人类遗传性疾病, 由于特定基因的缺失或突变, 使体内多种器官功能逐渐消退, 例如导致呼吸和行动困难以及提早死亡。囊性纤维化疾病是由于编码囊性纤维化跨膜传导调节因子(cystic fibrosis transmembrane conductance regulator, CFTR)的基因发生突变, 蛋白中Phe508残基缺失, 使CFTR蛋白折叠、运输和膜稳定性发生变化。CFTR构成了蛋白激酶A (PKA)调控的阴离子通道, 调控许多器官上皮细胞的氯离子和碳酸氢根离子的转运, 因而CFTR基因变异往往影响全身的机能。囊性纤维化是一类凶恶的罕见病, 没有有效的治疗药物。
2 苗头化合物 2.1 研发目标和活性评价Vertex公司的研发目标是治疗囊性纤维化可口服的小分子药物, 作用机制是通过恢复变异的CFTR通道功能, 达到缓解和治疗效果。筛选和评价化合物活性的方法是通过两种互补的机制影响CFTR的功能, 分别是增强和纠正CFTR功能。增强功能是化合物通过促进CFTR离子通道开启的几率, 增加离子的流动; 方法是将NIH-3T3小鼠成纤维细胞表达人缺失Phe508的CFTR蛋白, 通过毛喉素(forskolin, 天然二萜, 为cAMP酶激活剂)刺激, 在受试物的作用下, 使Cl-离子流出, 从而改变了膜电位, 用荧光法测定膜电位的变化的强弱, 表征受试物的活性。化合物的纠正功能是改善缺失Phe508的CFTR的加工和向细胞表面的传输, 从而提高CFTR介导氯离子的分泌。方法是将成纤维细胞与受试物温孵16 h, 按上述方法测定膜电位的变化。
2.2 高通量筛选获得苗头化合物为了发现苗头化合物, 对228 000个化合物进行了高通量筛选, 筛选方法是用上述促进离子通道的开启功能的模型(Goor FV, Hadida S, Grootenhuis PDJ, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Am Sci U S A, 2009, 106: 18825-18830), 结果发现了化合物1, EC50为2.1 μmol·L-1。证明化合物1是通道蛋白的促进剂, 而不是CFTR功能的激活剂。另外, 用患者的支气管上皮细胞(缺失Phe508-CFTR蛋白)测定活性, EC50为1.5 μmol·L-1, 但刺激Cl-离子分泌的EC50为12.2 μmol·L-1, 再一次证实激活CFTR的功能较弱。
从结构上分析化合物1, 母核喹啉酮可发生互变异构成为4-羟基喹啉, 羟基与酰胺的氧可形成分子内氢键而稳定化, 所以1存在有两种结构形态, 这在优化和结构变换中是需要考虑的问题。

|
为了研究构效关系和优化结构, 研发者将1分解成两个部分, 即羰基左侧的喹啉酮和右侧的胺片段, 分别对着两个片段加以变换。喹啉酮结构变换所合成的化合物列于表 1, 评价模型是对缺失Phe508-CFTR的NIH-3T3细胞促进通道开启的活性。
| Table 1 Compounds that convert quinolinone fragments |

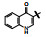
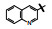

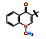
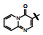
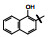
构效关系简析如下: ①化合物2是1的烯醇式去羟基物, 无取代的喹啉环使活性下降; ②去除并合的苯环(3)失去活性, 提示该片段要有一定的亲脂性; 4和5失去了N1上的氢原子而无活性, 说明N-H的重要性, 推测作为氢键给体参与结合作用; ③萘酚基本保持活性, 推测1的烯醇式也可能是与蛋白结合的结构。
3.2 二苄基胺的变换为优化右侧的二苄基胺, 设计合成了大约70多个化合物, 包括伯、仲、叔胺、芳香胺和杂环胺等, 表 2列出有代表性化合物的结构与活性。
| Table 2 Compounds that change amine structure |
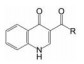
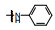

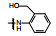

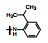
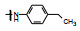



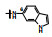
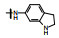
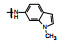
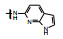
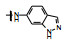

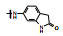
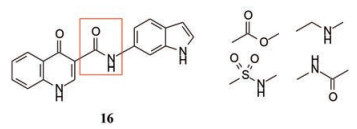
表 2的构效关系分析如下: ①苯胺(7)、2, 6-二乙基苯胺(10)、4-乙基苯胺(12)和β-萘胺(14)等的活性与1相似; ②苯胺的邻位有一个烷基(8和11)或对位有支链烷基(13), 可使活性提高5~10倍; ③化合物17为吲哚晽结构, 活性是1的3倍, 杂环结构是一个优化的生长点; ④化合物16连接的是6-氨基吲哚, EC50 = 0.1 μmol·L-1, 比化合物1提高20倍。若连接成5-氨基吲哚(15)或将16的N1甲基化(18)活性下降60倍。提示16的两个NH对结合的重要性。
对高活性的化合物16用60多种靶标评价其脱靶作用, 除对GABAA苯并二氮卓受体(氯通道)有较强的活性外(IC50 = 0.1 μmol·L-1), 都没有呈现活性, 表明16对缺失Phe508-CFTR蛋白的细胞有良好的选择性。然而16的溶解度较小, 可能的原因是4-酮基与NH发生氢键结合, 形成平面性分子, 进而分子间氢键结合使排布紧密, 高晶格能导致溶解度降低。X-射线分析16的晶体结构证实了这种推断。图 1是化合物16的晶体X-射线衍射测定的结构。

|
Figure 1 X-ray structure of compound 16 |
化合物16作为新一轮的先导物进行优化, 首先考察连接两片段的酰胺基的变换对活性的影响。为此设计了由酯基、磺酰胺基、亚甲胺基和胺酰基作为连接基的化合物, 结果表明活性都非常弱, 在30 μmol·L-1浓度下活性为1%~19%, 提示-CO-NH-连接基不能变动, 即使反向连接成-NH-CO-的化合物也没有活性, 说明这个连接基是不能变动的。
|
由于化合物8和13分别在2位和4位引入烷基, 显著提高了活性, 所以在吲哚环的3位或5位(相当于8和13的2和4位)引入烷基。合成的烷基化合物列于表 3。结果提示, 3-乙基吲哚(23)与16的活性相同, 5-乙基(25)则提高了5倍; 3-或5-叔丁基(24, 26)提高了10倍, 提示高亲脂性和空间占位性有利于活性。
| Table 3 Indole ring contains alkyl compound |

化合物17是二氢吲哚环, 剖裂2, 3位的饱和碳键而成为含开链烷基的苯胺化合物, 设计合成了表 4的化合物。27的苯环上没有烷基取代, 活性比16降低近70倍, 随着引入大尺寸和叉链烷基, 活性则提高, 例如4-叔丁基取代的化合物24活性与16相同。再次表明酰胺的对位含有叉链烷基有利于活性。
| Table 4 Ring-opened amine compound |
化合物32的结构简化为3-氨基4-叔丁基取代, 显示了高活性, EC50为0.1 μmol·L-1, 所以固定4-叔丁基, 探索变换3-氨基对活性的影响。表 5列出的化合物表明, 氨基被酰化(33和34)活性显著下降, 用氨甲基(35)、羧基(37)和磺酰氨基(38)置换的化合物也使活性减弱, 而用羟甲基(36)或没有取代基(39)或氟代(40)仍保持活性, EC50为0.1 μmol·L-1。
| Table 5 The activity of the compound that changes the 3-amino group |

3-羟基化合物41显示了意外的高活性, 比16提高了40倍。羟基为氢键给体(也是接受体), 体积较小, 可能是高活性的原因。这也再现了药物化学中苯酚与吲哚环互为等排体的概念(在多巴胺受体激动剂的设计中已有应用) (Asselin AA, Humber LG, Voith K, et al. Drug design via pharmacophore identification. Dopaminergic activity of 3H-benz[e]indol-8-amines and their mode of interaction with the dopamine receptor. J Med Chem, 1986, 29: 648-654)。用患者支气管上皮细胞(缺失Phe508-CFTR蛋白)测定化合物41的活性, EC50为5 nmol·L-1, 比化合物1提高了300倍。
4.5 苯环的6位变换优化至此, 3-羟基-4-叔丁基苯成为优化选择的片段。6位是个需要探索的位置, 因为前述的8、11和24等化合物的酰胺基邻位的烷基取代有增强活性的效应, 对应于此处的6位。故而合成了表 6的化合物。6位用F、CF3或叔丁基取代都显示高活性。结果表明6位有或没有取代基、以及不同性质的取代基都有很强的体外活性。
| Table 6 6-Position transformation of 3-hydroxy-4-tert-butylbenzene |
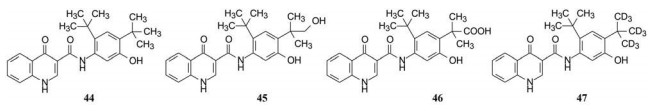
对优化出的吲哚系列化合物24和26和苯酚系列的41~44作多参数评价, 以确定候选化合物。表 7列出了这些化合物对患者支气管上皮细胞(缺失Phe508-CFTR蛋白)的体外活性(更接近患者细胞的体外模型)和大鼠的药代动力学性质。结果表明, 对支气管上皮细胞的抑制活性都在亚纳摩尔的高活性水平。化合物44静脉注射大鼠的药代显示的清除率和半衰期优于其他化合物。
| Table 7 Multi-parameter evaluation of compounds 24, 26 and 41-44. * In vitro activity measured with patient bronchial epithelial cells (without Phe508-CFTR protein) |
进而试验了44对160个蛋白的作用, 高浓度下未见显著作用, 表明没有脱靶作用, 其中包括对hERG离子通道无抑制活性, 提示对心脏的不良反应的风险较低。在20 μmol·L-1浓度下44对与药物代谢相关的CYP酶无抑制作用。
化合物44另一个重要作用是对双突变(F508缺失和G511D突变)的人支气管上皮细胞门控通道有较高的活性, 也提高了细胞对氯离子的分泌活性, EC50 = 236 nmol·L-1。所有这些性质都提示44有良好的开发前景, 因而用小鼠、大鼠、犬和猴经灌胃和静脉注射途径系统地评价了药代性质, 表明有良好的口服生物利用度和较长的半衰期(数据省略)。从而确定为候选化合物, 定名为依伐卡托(ivacaftor), 经临床前和临床研究, 2015年7月2日美国FDA批准上市, 可单一用药, 或与鲁玛卡托(lumacaftor)合用, 治疗囊性纤维化患者, 可显著改善囊性纤维化病人的肺功能。
6 依伐卡托的代谢转化和氘代物的研制依伐卡托为口服用药, 每日两次。体内外研究表明, 依伐卡托可被CYP3A4和3A5氧化代谢成M1 (45)和M6 (46)而失活(Schneider EK, Reyes-Ortega F, Wilson JW, et al. Development of HPLC and LC/MS-MS methods for the analysis of ivacaftor, its major metabolites and lumacaftor in plasma and sputum of cystic fibrosis patients treated with orkambi or kalydeco. J Chromatog B, 2016, 1038: 57-62)。为降低依伐卡托的代谢速率, Concert公司利用其DCE (氘代化学实体)平台技术将4位叔丁基变换为氘代叔丁基, 代号为CPT-656 (47)。由于C-D键的化学稳定性强于C-H键, 动力学同位素效应表明, 降低了CTP-656的代谢速率, 提高了稳定性, 延长了在体内的暴露时间, I期临床研究表明, CTP-656口服每日一次可维持有效浓度。Concert以2.5亿美元转让给原研公司Vertex公司。这是一个基于氘代改善药代动力学的例子。
新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
囊性纤维化属于罕见病, 是高致死率的遗传性疾病, 从基因和分子水平上解析发病原因和机制, 可为创制针对性的治疗药物提供依据和靶标以及评价的模型。从高通量的随机筛选到依伐卡托的批准上市, 研发过程清晰地显示出药物化学方法与构效分析的作用。此外, 针对依伐卡托代谢转化的位点而设计的氘代物, 具有延长半衰期和降低用药频度的良好前景, 显示了最简单的电子等排置换的有用性。
(编者按)