 2020, Vol. 55
2020, Vol. 55



2. 西南大学生命科学学院, 重庆 400715
2. School of Life Science, Southwest University, Chongqing 400715, China
青蒿素是中国科学家从植物黄花中提取的一种倍半萜内酯化合物, 结构新颖独特, 但结构本身难于直接修饰。双氢青蒿素(DHA)是青蒿素的还原产物, 不仅保留了抗疟活性的必要母体结构, 而且显示了比青蒿素更为优良的药学性质, 更为重要的是提供了直接进行结构修饰的反应位点—C-12位的羟基。中国科学家对DHA的羟基进行甲基修饰, 成功获得第一个青蒿素类抗疟药物蒿甲醚(artemethere)[1]; DHA与丁二酸酐反应, 获得第二个青蒿素类抗疟药物—水溶性青蒿琥酯(artesunate, 图 1)[2], 进一步显示了C-12位羟基修饰的前景。

|
Figure 1 First-generation artemisinin antimalarials |
DHA第一代衍生物的成功开发, 吸引了全球众多药物工作者的参与, 结构各异的DHA衍生物被合成出来; 这些O-苷、C-苷和N-苷类衍生物(图 2), 有的显示优异的抗疟活性, 有的表现为抗炎、抗肿瘤、抗肺纤维化、抗病毒、抗菌增敏或抗结核作用等[3, 4]。这些研究, 不仅证实了DHA的C-12位可以进行各种类型的修饰, 而且表明修饰产物可能具有多种多样的生物活性。

|
Figure 2 Some types of dihydroartemisinin (DHA) derivatives |
氟喹诺酮类(FQs)药物是喹诺酮药物发展史上的里程碑。FQs具有较高的口服生物利用度、良好的药代动力学特性和优异的抗感染活性, 已广泛应用于呼吸道感染、胃肠道和泌尿道交叉感染、皮肤和软组织交叉感染、慢性骨髓炎以及性传播疾病等的治疗[5, 6]。进一步研究发现, FQs还具有多种非典型生物活性, 包括抗结核、抗肿瘤、抗人类免疫缺陷病毒(HIV)、抗疟疾和抗阿尔茨海默病[7]。FQs骨架是成药性最高的结构之一。2009年, 杨玉社研究团队[8]开发的FQs新药盐酸安妥沙星成功上市; 最近几年, 国内外仍有大量的FQs研究论文发表或者活性分子甚至先导分子发现[9, 10]。分析上市药物和文献中的活性分子发现, 保留FQs基本骨架, 对其C-7位杂环的改变或修饰最多也最为成功。基于DHA和FQs的可修饰位点及其成功案例, 本研究设计了图 3所示的分子模式。

|
Figure 3 Model A of target molecule |
为了验证研究的可行性, 选取第4代FQs中的明星分子克林沙星与简单的亚乙基和亚丙基连接, 获得了高活性的目标分子[11] (图 4)。

|
Figure 4 Highly bioactive molecules founded previously |
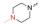
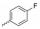
但是, 目标分子linker简单、FQs只选用了克林沙星。为了探索其他FQs及linker的可用性和新分子的生物活性, 本研究拟选取结构复杂的linker和多样性的FQs。考虑到分子的可连接性和FQs结构中XYZ的不同类型, 本研究尝试性地选择了如下9个FQs药物(图 5)。

|
Figure 5 Nine selected fluoroquinolone drugs in clinical |
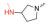
众所周知, Linker首先必须拥有连接功能, 其次尽可能满足分子量小、分子刚柔性适度、代谢片段无毒等基本要求。鉴于Linker是连接DHA的羟基和FQs的氨基, 那么Linker含有羟基和羧基具有优势。考虑到分子复杂、无毒以及结构可修饰等设想, 选定L-高丝氨酸为设计分子的Linker骨架: L-高丝氨酸含有羟基、氨基和羧基3个官能团, 其中羟基和DHA连接、羧基和FQs反应, 剩下一个可以修饰的氨基。有文献报道, 氨基被保护的L-高丝氨酸是合成非蛋白质氨基酸的重要手性源化合物, 也是合成某些药物(如抗癌物质刀豆氨酸和S-腺苷甲硫氨酸等)的重要中间体。为了调整分子的理化特性, 选择苄氧羰基(Cbz)、叔丁氧羰基(Boc)这两个特色氨基保护基对L-高丝氨酸的氨基进行修饰。作为尝试, 设计了两类目标分子(图 6)。

|
Figure 6 Designed target molecules |
基于目标分子模式, 本研究设计并合成了18个目标分子; 美国礼来制药公司(Eli Lilly and Company)进行生物活性测试, 得到多种生物活性。本文即报道上述研究结果。
结果与讨论 1 化学部分设计的目标分子TM1/TM2, 是双氢青蒿素与氟喹诺酮片段连接物, 结构由3部分组成, 因此有如下两条合成路线(合成路线1)。

|
Scheme 1 Designed target molecules |
氨基酸进行Boc或Cbz保护得到Linker分子IM1。Route 1是先将IM1与DHA成醚连接, 合成关键中间体IM2, 随后采用酰胺合成法连接沙星和IM2, 进而合成目标分子TM。此路线的优点是偶联片段大小相近, 有前期合成经验; 不足是沙星溶解性差, IM2价格相对较高不宜超量使用, 可能影响反应速度和反应收率。Route 2是先将沙星与IM1连接合成中间体IM2', 其后再和DHA偶联合成目标分子TM。由于DHA过氧桥键不稳定, DHA参与的反应越少越好; IM2'溶解性可能比沙星好, 有利于其与DHA偶联。Route 2看起来是一条不错的路线。但是本研究室前期研究发现, DHA与含羟基的小分子能够形成醚键, 但与含羟基的复杂分子反应难度较大, 据此推测, 由DHA和中间体IM2'连接形成醚键的反应难度可能很大甚至不发生反应; 此外通过Route 2制备TM, 从头到尾没有一个共同的中间体, 合成工作量很大, 不符合有机合成的基本原则。初步实验证实, IM2'与DHA成醚反应不能进行。因此选择Route 1为TM1/TM2的合成路线(合成路线2)。

|
Scheme 2 Synthetic routes of TM1/TM2 (i) a. CbzCl, Na2CO3; b. (Boc)2O, Na2CO3; (ii) DHA, BF3·Et2O, Et2O; (iii) FQs, t-BuCOCl, DIPEA, DCM, -3 ℃ |
L-高丝氨酸分别与Cbz-Cl和Boc2O反应合成IM1-1和IM2-1, 收率超过90%。DHA与高丝氨酸中的羟基反应, 未见文献报道, 作者发现用BF3·Et2O做催化剂、Et2O做溶剂, 在-5 ℃控温反应最好, 收率可达55%左右。从FQs获取目标分子TM1及TM2的反应是酰胺形成反应。实验发现, 采用常规的多肽偶联法, 反应慢杂质多, 产物还不易纯化, 主要原因在于DHA的不稳定性和FQs的低溶解性。利用本研究室长期开展多肽合成和酰胺类化合物合成的丰富经验, 发现特戊酰氯是合成目标分子的最好试剂, 通过条件摸索, 较为顺利地合成了目标分子, 结果见表 1; 目标化合物的结构经1H NMR、13C NMR和HRMS确证, 具体数据见实验部分。
| Table 1 Experimental results of TM1/TM2. *Yield (%) refers to the yield of step iii |
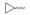




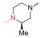

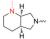

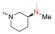

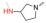

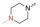
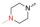
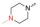
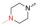
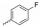
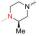
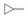
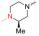


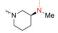
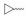
实验发现, 偶联反应的最适温度为-3 ℃。当FQ为克林沙星、环丙沙星、依诺沙星、诺氟沙星和沙拉沙星(TM1-1~TM1-5, TM2-1~TM2-5)时, TM1系列的反应时间为5.0~5.5 h, 产物收率44%~83%, TM2在相同条件下反应更快(3.5~4.0 h)且收率普遍更高(68%~85%)。当FQ为加替沙星、洛美沙星、莫西沙星和巴洛沙星(TM1-6~TM1-9, TM2-6~TM2-9)时, TM1的反应时间为3.0~3.5 h, 收率24%~74%; TM2的反应时间4.2~5.3 h, 收率42%~59%。由于洛美沙星和巴洛沙星反应效果不好, 因此TM1-7、TM1-9、TM2-7和TM2-9的收率较低。TM2的收率整体高于TM1, 可能与保护基的种类及产物的溶解性有一定的关系, 也与有些杂质需柱色谱纯化才能除掉有关。
2 生物活性评价 2.1 抗结核活性与理化性质的关系结核病(tuberculosis, TB)是结核分枝杆菌感染导致的重大传染病, 发病率和死亡率很高。结核病的治疗主要依赖化学药物。单一化学药物, 无论是新药还是传统老药, 长期用药后都会出现耐药性。为了克服耐药性, 一般采用联合用药和长期用药的策略。为了保证有药可用, 发现新型药物始终是医药界的追求。
FQs药物是耐药结核病的核心治疗药物。青蒿素类药物是目前治疗疟疾的最好药物。为了研究目标分子的生物活性, 首先进行了抗结核活性测试。活性测试在美国礼来制药公司的Open Innovation Drug Discovery program (OIDD)平台进行。开展TM1/TM2系列分子对非复制状态(hTHP-1)和复制状态(H37Rv)的结核分枝杆菌的抑制活性测试, 包括抑制率(5 μmol·L-1和20 μmol·L-1两个浓度)、半抑制浓度(IC50)以及细胞毒性(hTHP-1 Cytot)测试。此外, 利用美国礼来制药公司提供的Plexus软件, 计算了目标化合物的理化性质clogP及分子参数TPSA和Fsp3 (统称“分子参数”, 见表 2), 并讨论了化合物的抗结核活性与分子参数之间的关系。
| Table 2 Anti-TB activity and parameters of target molecules TM1/TM2. "/": Not calculated or determined |
从表 2看出, 在5 μmol·L-1测试浓度时, 几乎所有目标化合物对非复制状态的结核分枝杆菌THP-1的抑制率(45.69%~92.95%)比DHA (7.7%)显著提高:抑制率在80%以上有5个化合物, 其中TM2-5达到了92.95% (IC50 = 0.72 μmol·L-1); IC50在0.28~1.70 μmol·L-1, IC50 < 0.50 μmol·L-1的分子就有8个。在20 μmol·L-1测试浓度时, 有3个化合物对于复制状态的结核分枝杆菌H37Rv的抑制率高于50%, 特别是TM2-3达到了62.7%。构效关系分析可以看出, TM2 (Boc保护)较TM1 (Cbz保护)系列化合物具有更好的抗结核活性, 含有加替沙星(TM2-6/TM1-6)、沙拉沙星(TM2-5/TM1-5)的分子对非复制状态的结核分枝杆菌的抑制率都很好(>80%), 含有加替沙星(TM2-6/TM1-6)、克林沙星(TM2-1/TM1-1)的分子对H37Rv结核分枝杆菌的抑制率也不错(强于DHA)。此外, 测试的分子对两种细胞的抑制活性强度几乎不一致, 可能分子对复制与非复制细胞的抑制机制差异较大。
表 2中hTHP-1 Cytot.表达分子对非复制结核菌株的细胞毒性, 通常IC50值越大分子对细胞的毒性越低。所测试的分子, 其IC50值在0.46~6.22 mg·L-1, 说明这些分子对hTHP-1细胞毒性相差较大; 遗憾的是, 对hTHP-1细胞抑制率高于80%的化合物的细胞毒性虽不是最强但整体不低。
分子的理化性质及参数(表 2)也是先导化合物确定时必需考虑的因素。Fsp3是sp3杂化碳数与总碳数的比值, 是描述化合物不饱和度的参数[12]。有研究表明, 高的Fsp3数值代表着分子拥有更多可旋转的碳, 意味着分子柔性更好、空间适应性更强, 以致分子中原有的苯环可以更好地与蛋白质残基中的芳香环产生π-π堆积作用[13], 从而与受体蛋白更好地结合。从表 2看出, 化合物Fsp3值大, 其对H37Rv的抑制活性可能大(如TM2-3/2-1/2-6/1-6)也可能小(如TM2-9/2-8/2-2), 对hTHP-1的抑制活性与对H37Rv的活性大致相同, 虽然TM2-5的Fsp3不大(0.59)但其抑制活性最高(92.95%)、TM1-5的Fsp3不大(0.49)但其抑制活性不错(82.51%), 表明Fsp3值和活性没有必然联系。
clogP是衡量分子脂溶性的重要指标。抗结核药物需要通过由磷脂双分子层组成的人体细胞膜和结核分支杆菌的高脂质含量的细胞壁, 因此脂溶性直接影响着药物的透膜性、吸收、分布、代谢、外排和毒性。针对结核分枝杆菌, 需要脂溶性较高的分子。本研究设计的18个分子的clogP≥5, 与目前结核分子设计的趋势一致; 从表 2可以看出, TM1的clogP值整体上高于TM2, 但是TM1系列分子的抑制活性, 无论对hTHP-1还是H37Rv, 并非都好于TM2, 虽然活性好的分子一般还是具有较高的clogP值。然而, clogP值最大(7.37和6.70)的两个分子TM1-5和TM2-5, 抑制hTHP-1的活性是最强的, clogP值最小(5.20和5.31)的两个分子TM2-4和TM2-2, 其抗结核菌活性确实是最低的, 表明活性与clogP值一般成正比。
TPSA (拓扑极性表面积)是分子中极性原子的表面积总和, 它是预测药物吸收能力的一个参数, 常作为药物透膜性的评价指标[14]。对于口服药物的TPSA应小于140, 有助于口服吸收, 但抗菌药物不同于一般药物, 一般需要较大的TPSA。表 2可以看出, 活性好的分子, 一般来说TPSA值都较大, 但并非TPSA值大的分子就有好的活性。
本研究所得分子的生物活性及分子参数, 对后续研究应该具有一定的指导意义。
2.2 降血脂靶点PCSK9抑制活性前蛋白转化酶枯草溶菌素9 (proprotein convertase subtilisin/kexin type 9, PCSK9)是2003年发现的一个脂质代谢调节蛋白。大量研究发现, PCSK9能介导低密度脂蛋白受体降解, 调节血浆低密度脂蛋白胆固醇水平。因此, 抑制或降低PCSK9水平的治疗方法可有效治疗高胆固醇血症, 已成为高胆固醇血症研究的热点[15-17]。针对PCSK9的生理特点及作用机制[18], PCSK9抑制剂可分为以下3种:抑制它与低密度脂蛋白胆固醇结合的单克隆抗体或小分子多肽; 抑制PCSK9合成的反义寡核氨酸或小干扰RNA (siRNA); 阻断PCSK9在内质网中的自身裂变的小分子抑制物。
目标化合物TM1/TM2系列的PCSK9抑制活性由美国礼来制药公司OIDD平台按照如下流程进行测试:首先进行单浓度初筛, 采用AlphaLisa方法测试化合物对肝癌细胞Huh7分泌PCSK9的百分抑制率, 并采用CellTiter-Glo试剂测试化合物对肝癌细胞Huh7活力的影响(细胞毒性); 对初步筛选出的潜力分子进行多浓度测试; 然后进行复筛, 采用ELISA法检测24 h处理后Huh7细胞ApoA-I蛋白表达抑制。
由表 3发现, 在测试浓度下, 有13个化合物的PCSK9抑制率超过72%, 其中TM1-3、TM1-7、TM2-9、TM2-8、TM2-5的抑制率高于80%, 且TM1-3达到了92.30%, 显示良好的抑制活性。从结构上看, 喹诺酮环8-位含有甲氧基的分子, 都显示较好的抑制活性。这是首次发现L-高丝氨酸连接的双氢青蒿素-氟喹诺酮偶联物具有PCSK9抑制活性。遗憾的是, 活性较好的分子对肝癌细胞Huh7的细胞毒性可能较大。
| Table 3 PCSK9 inhibitory activity of TM1/TM2 |
为寻找青蒿素-氟喹诺酮连接物的高活性低毒性的先导分子, 采用美国Simulations Plus公司开发的ADMET-Predictor 7.0软件预测TM1和TM2系列目标化合物的毒性, 包括心脏毒性(TOX hERG)、致癌毒性(TOX BRM Rat and BRM Mouse)、致突变性(TOX MUT Risk)和综合毒性(TOX Risk), 给出毒性风险系数评估, 进而判断分子成药性。
TOX-BRM-Rat预测化合物的致癌性, 动物模型为大鼠, 规定TD50 ≥ 4 mg·kg-1·d-1为安全值, 从图 7 (左)可以看出, 合成的18个化合物TD50 ≥ 20 mg·kg-1·d-1, 应该没有致癌性。TOX-BRM-Mouse也是预测化合物的致癌性, 但动物为小鼠, 规定TD50 ≥ 25 mg·kg-1·d-1为安全值, 从图 7 (右)可以看出, 所有分子均无致癌性。

|
Figure 7 Prediction results of carcinogenic toxicity of TM1/TM2 (the left and right animal models are rats and mice, respectively) |
从图 8可以看出, 目标分子的心脏毒性(TOX hERG) ≤ 4.51, 而心脏毒性的规定值应小于6.0, 预测无心脏毒性。TOX-MUT-Risk是预测化合物在不同的沙门菌属中的致突变可能性的综合评价。目前大多数药物分子满足TOX MUT Risk ≤ 2的要求, 所有目标分子的TOX MUT Risk数值为1, 表明它们均无致突变性。TOX-Risk对于毒性的综合打分应当≤ 3.0, 该风险系数越大, 表示化合物的毒性性质越不理想。图 8显示, TM2系列分子的毒性风险较低且低于TM1。

|
Figure 8 The line chart of TOX hERG, TOX risk and TOX MUT risk of the TM1/TM2, and the ordinate represents the predicted value |
综上所述, TM2系列分子的安全性几乎都高于TM1, 可能与这些分子中的氨基酸保护基有关, 表明分子的安全性可以通过结构修饰调整, 这为后续分子的设计和改构指明了方向。
3 结论本研究采用活性拼接原理, 将DHA与9个氟喹诺酮药物分子通过高丝氨酸连接, 设计并合成了18个新分子, 收率24%~85%。目标分子结构得到1H NMR、13C NMR、HR-MS的确证。测试了所有目标化合物对MTB的体外活性和细胞毒性, 发现目标化合物对非复制状态结核分枝杆菌(hTHP-1)的抑制活性好于复制状态的结核分枝杆菌(H37Rv), 其中TM2-5对hTHP-1的抑制率高达92.5%;同一化合物对这两种MTB菌株的抑制活性差别很大。本研究首次发现, DHA与FQs偶联物对PCSK9具有抑制活性。软件计算发现, 几乎所有目标分子都没有致癌性、致突变性、心脏毒性。这些研究结果, 为后续研究奠定了基础。
实验部分 1 目标化合物的合成克林沙星、沙拉沙星(郑州克尔泰生化科技有限公司, >95%); 诺氟沙星、环丙沙星、洛美沙星、加替沙星、莫西沙星(成都爱斯特贸易有限公司, AR); 依诺沙星、巴洛沙星、氯甲酸苄酯(Cbz-Cl) (上海达瑞精细化工有限公司, AR); 双氢青蒿素(重庆华立武陵山制药有限公司, AR); 46.5%三氟化硼乙醚溶液(BF3·Et2O) (上海晶纯试剂有限公司, AR); L-高丝氨酸(L-HoSer)、N-苄氧羰氧基丁二酰亚胺(Z-OSu)、二碳酸二叔丁酯(Boc2O) (成都凯泰新技术有限责任公司, AR); 其余试剂均为市售化学纯或分析纯产品, 未经纯化直接使用。
磁力搅拌低温恒温水槽(PSL-1810, 上海爱朗仪器有限公司); 熔点测定仪(X-6, 北京福凯仪器有限公司); 自动旋光仪(WZZ-2S, 上海精密科学仪器有限公司); 核磁共振仪(600 MHz, Bruker, 瑞士, TMS为内标); 高分辨质谱仪(HR-MS) (Varian7.0T, Varian, USA)。
1.1 中间体IM1-1/IM2-1的合成于100 mL反应瓶中加入L-HoSer (1 mmol)、冷却的饱和Na2CO3溶液3 mL, 冰浴搅拌溶解; 滴加Cbz-Cl/Boc2O (1.5 mmol)的丙酮溶液2 mL; 室温搅拌反应, 必要时补加Na2CO3溶液调节pH>9; TLC-紫外荧光及茚三酮显色法监测反应进程。反应结束后, 加水15 mL, 在pH>9时用乙醚(10 mL×2)萃取, 收集水相, 1.5 mol·L-1盐酸调节pH至3~4, 乙酸乙酯(10 mL×3)萃取, 合并有机相, 无水Na2SO4干燥, TLC-茚三酮显色法检查纯度, 减压蒸干, 真空干燥, 得中间体IM1-1/IM2-1, 收率分别为94%、92%, 低温保存。
1.2 中间体IM1-2/IM2-2的合成于100 mL反应瓶中依次加入IM1-1/IM2-1 (1.0 mmol)及Et2O 3 mL, 室温搅拌溶解, 控温加入BF3·Et2O 0.2 mL, 搅拌0.5 h, 加DHA (1.5 mmol), 继续控温搅拌反应, TLC-紫外荧光和磷钼酸显色法监测反应进程。反应结束后, 加Et2O 15 mL, 饱和NaHCO3溶液(10 mL×3)萃取, 合并水相, 用1 mol·L-1盐酸调节pH至3~4, EtOAc (10 mL×3)萃取, 合并有机相, 饱和NaCl溶液(10 mL×3)洗涤, 无水Na2SO4干燥, 减压蒸干得到粗品; 柱色谱纯化, 收集洗脱液, 减压蒸干, TLC-紫外荧光和磷钼酸显色法检查纯度, 真空干燥, 得中间体IM1-2/IM2-2, 收率分别为55%、59%, 低温保存。
1.3 目标化合物TM1/TM2系列的合成于100 mL反应瓶中依次加入IM1-2/IM2-2 (1.2 mmol)和二氯甲烷(DCM) 3 mL, -3 ℃搅拌溶解, 顺次加入N, N'-二异丙基乙胺(DIPEA) (1.5 mmol)和特戊酰氯(1.5 mmol), -3 ℃搅拌反应0.5 h, 分别加入FQ (克林沙星、诺氟沙星、环丙沙星、沙拉沙星、莫西沙星、洛美沙星、加替沙星、依诺沙星或巴洛沙星)(1 mmol), 继续在-3 ℃搅拌反应, TLC监测反应进程。反应结束后, 减压抽滤, 滤饼用DCM (2 mL×3)洗涤, 合并洗液及滤液, 加DCM 20 mL, 依次用饱和NaHCO3溶液(15 mL×2)、5%柠檬酸水溶液(15 mL×2)、饱和NaCl溶液(10 mL×2)洗涤, 无水Na2SO4干燥, 减压蒸干得到粗品; 柱色谱纯化[PE:EA=1:1 (体积比)], 收集洗脱液, 减压蒸干, 乙醚重结晶, TLC-紫外荧光和磷钼酸显色法检查纯度, 真空干燥, 得目标化合物TM1/TM2。
TM1/TM2系列化合物的表征数据如下:
TM1-1:淡黄色固体, mp 159~161 ℃,
TM1-2:淡黄色固体, mp 162~164 ℃,
TM1-3:淡黄色固体, mp 191~193 ℃,
TM1-4:淡黄色固体, mp 132~134 ℃,
TM1-5:淡黄色固体, mp 158~160 ℃,
TM1-6:淡黄色固体, mp 152~154 ℃,
TM1-7:淡黄色固体, mp 160~162 ℃,
TM1-8:淡黄色固体, mp 164~166 ℃,
TM1-9:淡黄色固体, mp 165~167 ℃,
TM2-1:淡黄色固体, mp 132~135 ℃,
TM2-2:淡黄色固体, mp 141~144 ℃,
TM2-3:淡黄色固体, mp 135~137 ℃,
TM2-4:淡黄色固体, mp 155~157 ℃,
TM2-5:淡黄色固体, mp 180~182 ℃,
TM2-6:淡黄色固体, mp 140~142 ℃,
TM2-7:淡黄色固体, mp 148~150 ℃,
TM2-8:淡黄色固体, mp 173~175 ℃,
TM2-9:淡黄色固体, mp 126~128 ℃,
测试工作由美国礼来制药公司的OIDD平台以及美国传染病研究所(Infectious Disease Research Institute, IDRI)的研究团队完成, 具体实验结果见表 2、3。测试方法参见文献[19-21]。
3 目标化合物的毒性预测[22-24]为寻找青蒿素-氟喹诺酮连接物的高活性低毒性的先导分子, 采用美国Simulations Plus公司开发的ADMET-Predictor 7.0软件计算目标化合物的心脏毒性、致癌毒性、致突变性和综合毒性, 同时也用美国礼来制药公司提供的Plexus软件, 计算了目标化合物的物理参数(表 2), 对需合成的目标分子先计算、再合成和测试活性。
致谢: 美国礼来制药公司和美国传染病研究所研究团队在活性测试方面提供的帮助。本学院老师在质谱和核磁共振测试中提供的支持和帮助。
作者贡献: 潘建芳负责部分实验, 论文的撰写、修改和校对; 孙晓丽负责分子设计、大多数化合物的合成, 文本校对; 范莉、唐雪梅、罗鹏负责少数分子合成, 文本修改、校对; 杨大成负责课题规划, 指导实验的设计、化合物的合成, 对外联系, 文本的修改。
利益冲突: 无利益冲突。
| [1] |
Li GQ, Li Y, Li ZL, et al. Artemisinin Antimalarials (青蒿素类抗疟药)[M]. Beijing: Science Press, 2015.
|
| [2] |
Lin AJ, Klayman DL, Milhous WK. Antimalarial activity of new water-soluble dihydroartemisinin derivatives[J]. J Med Chem, 1987, 30: 2147-2150. DOI:10.1021/jm00394a037 |
| [3] |
Jiang WW, Qian Y. Research progress on antibacterial activity of artemisinin and its derivatives[J]. China Pharm (中国药房), 2019, 30: 2003-2007. |
| [4] |
Zhang N, Liu Y, Li Y, et al. Enhancement of water solubility and anti-tuberculosis activities by a novel artemisinin derivative[J]. Acta Pharm Sin (药学学报), 2019, 54: 36-40. |
| [5] |
Zhang GF, Zhang S, Pan B, et al. 4-Quinolone derivatives and their activities against gram positive pathogens[J]. Eur J Med Chem, 2018, 143: 710-723. DOI:10.1016/j.ejmech.2017.11.082 |
| [6] |
Hu YQ, Zhang S, Xu Z, et al. 4-Quinolone hybrids and their antibacterial activities[J]. Eur J Med Chem, 2017, 141: 335-345. DOI:10.1016/j.ejmech.2017.09.050 |
| [7] |
Wang AP, Feng LS, Liu ML, et al. Recent research progress of fluoroquinolones antibacterials[J]. World Notes Antibiot (国外医药抗生素分册), 2019, 40: 171-179. |
| [8] |
Yang ZY, Sun DM. Antofloxacin hydrochloride, the first innovative fluoroquinolone drug in China, was successfully developed[J]. Prog Pharm Sci (药学进展), 2009, 33: 435-436. |
| [9] |
Sriram D, Aubry A, Yogeeswari P, et al. Gatifloxacin derivatives:synthesis, antimycobacterial activities and inhibition of mycobacterium tuberculosis DNA gyrase[J]. Cheminform, 2006, 16: 2982-2985. |
| [10] |
Carta A, Palomba M, Paglietti G, et al. [1, 2, 3] Triazolo[4, 5-h]quinolones. a new class of potent antitubercular agents against multidrug resistant mycobacterium tuberculosis strains[J]. Bioorg Med Chem Lett, 2007, 17: 4791-4794. DOI:10.1016/j.bmcl.2007.06.064 |
| [11] |
Zhou FW, Lei HS, Fan L, et al. Design, synthesis, and biological evaluation of dihydroartemisinin-fluoroquinolone conjugates as a novel type of potential antitubercular agents[J]. Bioorg Med Chem Lett, 2014, 24: 1912-1917. DOI:10.1016/j.bmcl.2014.03.010 |
| [12] |
Lovering F, Bikker J, Humblet C. Escape from flatland:increasing saturation as an approach to improving clinical success[J]. J Med Chem, 2009, 52: 6752-6756. DOI:10.1021/jm901241e |
| [13] |
Veber DF, Johnson SR, Cheng HY, et al. Molecular properties that influence the oral bioavailability of drug candidates[J]. J Med Chem, 2002, 45: 2615-2623. DOI:10.1021/jm020017n |
| [14] |
Veber DF, Johnson SR, Cheng HY, et al. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties[J]. J Med Chem, 2002, 45: 2615-2623. DOI:10.1021/jm020017n |
| [15] |
Cao A, Wu M, Li H, et al. Janus kinase activation by cytokine oncostatin M decreases PCSK9 expression in liver cells[J]. J Lipid Res, 2011, 52: 518-530. DOI:10.1194/jlr.M010603 |
| [16] |
Li H, Dong B, Park SW, et al. Hepatocyte nuclear factor lalpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine[J]. J Biol Chem, 2009, 284: 28885-28895. DOI:10.1074/jbc.M109.052407 |
| [17] |
Horton JD, Cohen JC, Hobbs HH. PCSK9:a convertase that coordinates LDL catabolism[J]. J Lipid Res, 2009, 50: 172-177. DOI:10.1194/jlr.R800091-JLR200 |
| [18] |
Desai NR, Kohli P, Giugliano RP, et al. AMG145, a monoclonal antibody against proprotein convertase subtilisin kexin type 9, significantly reduces lipoprotein(a) in hypercholesterolemic patients receiving statin therapy:an analysis from the LDL-C assessment with proprotein convertase subtilisin kexintype 9 monoclonal antibody inhibition combined with statin therapy (LAPLACE)-thrombolysis in myocardial infarction (TIMI) 57 trial[J]. Circulation, 2013, 128: 962-969. DOI:10.1161/CIRCULATIONAHA.113.001969 |
| [19] |
Liu J, Ren ZH, Fan L, et al. Design, synthesis, biological evaluation, structure-activity relationship, and toxicity of clinafloxacin-azole conjugates as novel antitubercular agents[J]. Bioorg Med Chem, 2019, 27: 175-187. DOI:10.1016/j.bmc.2018.11.035 |
| [20] |
Ollinger J, Bailey MA, Moraski GC, et al. A dual read-out assay to evaluate the potency of compounds active against mycobacterium tuberculosis[J]. PLoS One, 2013, 8: e60531. DOI:10.1371/journal.pone.0060531 |
| [21] |
Basak A, Chen A, Majumdar S, et al. In vitro assay for protease activity of proprotein convertase subtilisin kexins (PCSKs):an overall review of existing and new methodologies[J]. Methods Mol Biol, 2011, 768: 127-153. |
| [22] |
Feng JZ, Liu HP, Fan L, et al. A serial of Mannich bases with anti-leukemia activity[J]. Acta Pharm Sin (药学学报), 2017, 52: 766-772. |
| [23] |
Huang M, Xu J, Fan L, et al. Design, synthesis and antidiabetic activities of 5-arylmethylenethiazolidine-2, 4-dione derivatives[J]. Acta Pharm Sin (药学学报), 2017, 52: 1287-1298. |
| [24] |
Liu JY, Su XY, Li HC, et al. Design, synthesis, and evaluation of novel L-phenylglycine derivatives aspotential PPARγ lead compounds[J]. Bioorg Med Chem, 2018, 26: 4153-4167. DOI:10.1016/j.bmc.2018.07.005 |