 2020, Vol. 55
2020, Vol. 55

作者贡献:牟伊是本文的第一作者, 负责综述参考文献的收集整理, 主要内容的撰写; 文帅负责文章图片的绘制和表格内容的整理; 高新星负责靶向谷胱甘肽部分的整理和修改; 张蕊负责ROS生物学特征部分的整理和修改; 王燕负责ROS与肿瘤部分的整理和修改; 姜正羽为本文的通讯作者, 负责综述框架设计, 综述内容把关, 稿件修改等工作。
利益冲突:本文的研究内容无任何利益冲突。


2. 中国药科大学药学院, 江苏省药物分子设计与成药性优化重点实验室, 江苏 南京 210009
2. Jiangsu Key Laboratory of Drug Design and Optimization, School of Pharmacy, China Pharmaceutical University, Nanjing 210009, China
活性氧(reactive oxygen species, ROS)的经典定义是氧的部分代谢产物, 主要包括超氧化物(O2•-)、过氧化氢(H2O2)、羟基阴离子(OH-)、羟基自由基(OH•)和次氯酸(HOCl)等[1]。ROS主要由线粒体有氧呼吸中的电子传递链和氧化还原酶(黄嘌呤氧化酶、脂氧合酶、细胞色素P450、NADPH氧化酶等)等细胞代谢过程产生。此外, 外源性刺激, 如压力应激、紫外线辐射、肿瘤化疗和放疗等也可刺激ROS生成[2, 3]。在生理浓度下, ROS起信号分子的作用, 调节细胞生长、细胞间黏附、细胞分化、衰老和凋亡; 在高浓度时, ROS能破坏细胞生物分子, 如蛋白质、DNA或RNA, 引起基因突变, 促进正常细胞的癌变或导致癌细胞的耐药[4]。
研究显示肿瘤细胞与正常细胞相比处于更高的氧化应激状态, 而肿瘤细胞之所以有更高水平的ROS主要与其代谢异常有关。一般而言, 肿瘤细胞利用各种防御机制来减弱ROS的有害影响, 使它们在ROS的产生和消除保持相对平衡, 这对于肿瘤细胞的存活至关重要[5]。其中一条重要机制是过度表达抗氧化酶, 适应性增强抗氧化能力以清除过高的ROS水平。肿瘤细胞可能处于更高的氧化应激状态环境, 因此通过小分子破坏氧化还原平衡的治疗方法能优先杀伤肿瘤细胞[6]。
1 ROS的生物学特性 1.1 ROS的种类与来源ROS是指含有氧原子的不稳定分子, 常见ROS主要包括超氧化物(O2•-)、过氧化氢(H2O2)、羟基阴离子(OH-)、羟基自由基(OH•)、次氯酸(HOCl)、一氧化氮(NO)以及过氧亚硝酸盐(ONOO-)等。其中, O2•-主要由NADPH氧化酶/黄嘌呤氧化酶通过单电子还原O2而生成, 或者由线粒体电子传递链(ETC)产生, 此外非偶联内皮型一氧化氮合酶(endothelial nitric oxide synthase, eNOS)也产生部分O2•- [7]。O2•-能被超氧化物歧化酶(superoxide dismutase, SOD)快速分解为H2O2。当NO存在时, O2•-还可与NO迅速反应, 形成高活性的ONOO-, 此过程比O2•-向H2O2的转化快3~4倍。H2O2又可以被髓过氧化物酶(myeloperoxidase, MPO)转变为高活性的HOCl, 该酶在中性粒细胞中大量表达。在Fe2+作用下, H2O2也可以通过Fentons反应转变成高毒性的OH•。过氧化氢酶(catalase)、谷胱甘肽过氧化物酶(glutathione peroxidase, GPX)或过氧化物还原酶(peroxiredoxins, Prx)可将H2O2代谢为H2O和O2[8]。图 1展示了ROS的产生和转化机制。

|
Figure 1 Sources of reactive oxygen species (ROS) and antioxidant defense system. Superoxide (O2•-) is produced by NADPH oxidase/xanthine oxidase-derived reduction of O2, uncoupled endothelial nitric oxide synthase (eNOS), or mitochondrial electron transport chain (ETC). O2•- can be rapidly metabolized to H2O2 by superoxide dismutase (SOD). H2O2 can be transformed to highly reactive HOCl by myeloperoxidase (MPO). H2O2 can also change to the highly toxic OH• in presence of Fe2+ by Fenton's reaction. H2O2 is scavenged to H2O and O2 by catalase, glutathione peroxidase (GPX), or peroxiredoxins (Prx) antioxidant enzymes |
由于ROS的高反应性, 极其容易对蛋白质/游离氨基酸、DNA/RNA和脂质造成损伤[3]。ROS可导致蛋白质、游离氨基酸及其残基的氧化, 氧化位点包括氨基酸α-C、蛋白质多肽链、疏水氨基酸残基等。一般而言, ROS可以通过α-酰胺化、二酰胺化、脯氨酸残基氧化、谷酰胺残基氧化、天门冬氨酰残基氧化裂解肽键[9]。ROS诱导的蛋白质氧化可能导致: ①芳香基团和脂肪族氨基酸侧链的羟基化, 芳香族氨基酸残基的硝化, 巯基的亚硝化, 甲硫氨酸残基的磺化, 芳香基团和基本氨基酸的氯化等; ②多肽链断裂形成蛋白交联聚合物; ③蛋白质官能团与多元不饱和脂肪酸或碳水化合物衍生物(糖基化/糖化氧化)氧化产物发生反应从而影响其正常生理功能[10]。此外大量研究报道了ROS通过损伤蛋白质, 促进癌细胞的产生、存活和转移。例如H2O2可以氧化脯氨酸羟化酶2 (prolyl hydroxylase domain protein 2, PHD2), 影响低氧诱导因子(hypoxia-inducible factor 1, HIF-1)的稳定, 而HIF-1与肿瘤血管生成和转移密切相关[11]。另外ROS还能通过蛋白激酶D1激活NF-κB, 或者通过激活表皮生长因子(epidermal growth factor receptor, EGFR)上调细胞增殖[12]。
ROS可通过单链断裂、点突变、错误编码、异常扩增等途径引起DNA损伤。ROS能够与DNA中的鸟嘌呤、胸腺嘧啶和脱氧核糖发生作用, 被氧化的嘌呤和嘧啶可能在基因修复过程产生DNA的错配[4]。由于DNA与组蛋白以染色质的形式构成复合物, ROS可以进一步影响DNA自由基加成物的氧化和还原反应[13]。ROS对DNA的损伤可能导致DNA-蛋白质交联的形成, 其在心脏和脑组织中的积累在衰老、癌症和神经退行性疾病中发挥重要作用[14]。ROS水平的升高还可能导致线粒体DNA突变, 最终导致电子传递链复合物Ⅰ活性损伤, 促进肿瘤转移。
脂质在细胞中的作用包括能量存储、信号转导、转运和组成细胞膜, 多种类型的脂质都易被ROS氧化[15]。ROS与脂质分子的反应可激活脂质过氧化自由基级联反应。ROS与脂肪酸侧链发生作用并夺取亚甲基碳的氢原子, 其中脂质中双键存在越多, 氢原子就越容易被夺去。氢原子离去后留下未配对的中心碳原子, 可能会产生高活性的碳自由基, 从而引起其他更多的氧化反应。在某些条件下, 从脂质侧链中夺去一个氢会引起连锁反应, 产生大量脂质过氧化产物。过量存在的ROS会使生物膜的脂质过氧化, 导致生物膜流动性下降、膜电位异常、细胞内容物的破裂和泄漏[16]。
2 ROS与肿瘤ROS在肿瘤中的作用是双面的。一方面ROS可激活多种氧化还原反应和信号通路, 比如MAPK通路、PI3K/AKT通路、NF-κB通路、Keap1-Nrf2-ARE通路等, 促进肿瘤发生、发展和转移; 另一方面, 当ROS过量存在时, 又可通过氧化应激机制引起细胞应激和损伤, 最终导致细胞死亡。根据ROS过量的严重程度和暴露时间不同, 氧化应激可以激活细胞存活或凋亡机制[17]。一般而言, 在低浓度下(通常是亚微摩尔浓度), ROS可作为有丝分裂原诱导正常细胞和癌细胞增殖和存活; 在中等浓度(< 10 μmol·L-1), ROS能诱导暂时或永久的细胞周期阻滞并促进细胞分化; 在更高浓度(10~30 μmol·L-1), ROS能破坏细胞生物大分子, 如脂质、蛋白质或核酸等, 引起基因突变, 从而促进正常细胞的癌变或肿瘤细胞的耐药[18]。
正常生理条件下, 氧化还原系统具有良好的协调性和平衡性。但是, 当存在明显的刺激时会破坏这种平衡, 如化疗或者辐射, 使ROS水平升高, 触发氧化应激, 产生细胞毒性或者激活细胞凋亡, 最终导致细胞死亡。然而, 大多数肿瘤细胞在内源性氧化应激存在时仍能存活, 使它们得以免于细胞凋亡并对多种化疗药物产生耐药性。肿瘤细胞具有各种防御机制来降低或消除ROS的不利影响, 使它们对ROS的产生和清除保持相对平衡, 其中一个重要的机制是其具有高度活化的抗氧化系统, 该系统主要包括谷胱甘肽(glutathione, GSH)、硫氧还蛋白还原酶(thioredoxin reductase, TrxRs)、Nrf2 (NF-E2-related factor 2)、锰依赖性超氧化物歧化酶(manganese superoxide dismutase, MnSOD)和过氧化氢酶等[19]。当细胞暴露于化疗、放疗等治疗后, 体外数据显示, 某些肿瘤细胞, 特别是MDR肿瘤细胞会上调ROS和抗氧化酶水平。肿瘤细胞长期暴露于高ROS水平可能会增加其耐药性。ROS介导的肿瘤耐药主要是由于对氧化信号敏感的转录因子的激活, 包括NF-κB、Nrf2、c-Jun和HIF-1α等, 上述转录因子的激活可以增强抗氧化系统的激活和促进细胞存活蛋白的表达[20] (图 2)。

|
Figure 2 ROS balance and their roles in regulating cancer cell proliferation and apoptosis. ROS is generated by cellular metabolism or induction by exogenous factors, playing a role in numerous reduction-oxidation reactions and signaling pathways that regulate cancer initialization and development, such as oxidative-stimulated MAPK, PI3K, NF-κB and Keap1-Nrf2-ARE pathway. |
肿瘤细胞的ROS水平较高, 抗氧化系统处于高度活化状态, 这种独特的生物化学特性为肿瘤药物的开发提供了潜在途径。通过小分子破坏氧化还原平衡的治疗方法能优先杀伤肿瘤细胞。同时, 由于耐药肿瘤细胞内的抗氧化酶表达水平和氧化应激状态环境通常更高, ROS调节药物对耐药肿瘤细胞的干预可能具有更好的治疗效应。近年来, 靶向肿瘤细胞内ROS调节的治疗药物研究取得了一定进展, 以下将根据其作用靶标的不同, 分类介绍各自的研究进展。
3.1 靶向线粒体电子传递链线粒体在细胞的存活和死亡过程中均起着十分重要的作用。肿瘤细胞因代谢更为活跃, 较正常细胞更容易受到线粒体的影响。由于线粒体有着调节/启动凋亡的关键作用, 靶向线粒体的活性分子被认为是一种潜在的肿瘤治疗策略[21]。线粒体电子传递链(electron transport chain, ETC)位于线粒体内膜, 由复合物Ⅰ~Ⅳ组成。在电子载体辅酶Q和Cyt c的帮助下, ETC可将电子从电子供体(通常为NADH), 从复合物Ⅰ和Ⅱ经Ⅲ传递到Ⅳ, 最终转移到电子受体(O2), 形成一个电化学质子梯度驱动ATP的合成[22]。该过程存在大量的电子流动, 其中小部分电子可能从ETC泄漏, 直接与O2结合, 形成O2•-, 这些自由基又可转化成其他ROS, 这使得ETC成为线粒体ROS的主要来源[23]。由于肿瘤细胞快速增殖, 某些肿瘤细胞中的ETC变得更加活跃以产生足够的ATP维持细胞增殖, 并通过ATP驱动的多种药物外排泵(包括P-gp)产生耐药性[24]。由于肿瘤细胞中ETC可能会产生更多的ROS, 其会上调某些抗氧化酶以适应更高的ROS水平。线粒体中产生的ROS可促进Cyt c从线粒体内膜分离, 这是诱导细胞凋亡的关键过程[25]。通过靶向ETC可阻断细胞内ATP的生成, 增加ROS的产生, 进而激活内源性凋亡途径, 促进肿瘤凋亡[26, 27]。图 3显示了在某些类型的肿瘤细胞中通过影响ETC增加ROS水平的化合物。

|
Figure 3 ROS modulators targeting mitochondria electron transport chain (ETC) |
伊利司莫(elesclomol)是一种ETC靶向化合物, 2008年FAD批准其为治疗转移性黑色素瘤的孤儿药, 目前正在美国进行转移性实体瘤和急性髓性白血病的临床试验(NCT00808418和NCT01280786)。伊利司莫在体内通过螯合铜离子转化为其活性形式, 这种复合物通过破坏ETC促进线粒体ROS的产生, 诱导氧化应激, 最终引起黑色素瘤细胞的死亡, 而这种效应会被线粒体靶向抗氧化剂Mito-Tempo部分逆转[28]。
鱼藤酮(rotenone)是一种从豆薯属植物中分离得到的异黄酮。它是ETC复合物Ⅰ抑制剂, 可增加肿瘤细胞中ROS的产生, 诱导肿瘤细胞凋亡。鱼藤酮在普通肝癌细胞SMMC-7721和对多柔比星耐药的SMMC-7721中均可产生类似的细胞毒性作用[29]。当鱼藤酮与紫草素联合使用时, 通过增加线粒体内的O2-水平, 协同增强胶质瘤细胞的凋亡, 而这一协同增效可被ROS清除剂MnTBAP预处理减弱[30]。鱼藤酮联合2-脱氧-D-葡萄糖(2-deoxy-D-glucose, 2-DG)使用, 可以显著升高人结直肠腺癌HT29和HCT116的ROS水平, 并且可以抑制HT29荷瘤小鼠的肿瘤生长[31]。
DT-010是丹参素和川芎嗪的缀合物, 可通过抑制ETC复合物Ⅱ, 促进乳腺癌MCF-7和MDA-MB-23细胞凋亡, 但是该效应可被N-乙酰半胱氨酸逆转[32]。在这两类细胞中, DT-010可以抑制线粒体呼吸, 以及损伤线粒体复合物Ⅱ的活性, 最终导致线粒体基本功能的抑制、线粒体膜电位下降、ATP产生减少、ROS水平升高。
3.2 靶向NADPH氧化酶(NOXs)NADPH氧化酶(NADPH-oxidase, NOXs)家族有7个亚型(NOX1、NOX2、NOX3、NOX4、NOX5、DUOX1和DUOX2), 不同亚型分布在不同的组织中。NOX家族的核心催化区均有相似的结构, 其基本功能是氧化O2生成的H2O2和O2- [33]。越来越多的证据显示, NOXs催化生成的ROS可能增加基因组不稳定的风险, 从而增加恶性肿瘤和耐药肿瘤的形成。例如, 在前列腺癌(NOX1)、胃癌和急性髓性白血病(NOX2)、黑色素瘤、恶性胶质瘤和甲状腺瘤(NOX4)的化疗中, NOXs均会产生一定的影响。无论是使用CRISPR-Cas9基因组编辑, 还是使用干扰RNA技术敲减NOXs, 都能显著降低肿瘤细胞的增殖和侵袭活性, 同时提高某些抗肿瘤药物的疗效。这些结果表明, NOXs可以成为潜在的抗肿瘤靶点。抑制NOXs可抑制过氧化氢和O2-介导的肿瘤细胞增殖, 而激活NOXs能增加癌细胞中活性氧的水平, 因此NOXs小分子抑制剂或激活剂均可能降低肿瘤细胞的生长[34]。本文讨论的作用于NOXs的化合物如图 4所示。

|
Figure 4 ROS modulators targeting NADPH-oxidase (NOXs) |
GKT137831是一种选择性NOX4抑制剂, 能抑制人肺动脉平滑肌细胞和人肺动脉内皮细胞中因缺氧产生的H2O2[35]。NOX4在NSCLC中高度表达, 促进葡萄糖代谢转移到糖酵解和磷酸戊糖途径以产生NADPH, 以增加谷胱甘肽的合成实现抗氧化, 最终促进肿瘤发展。GKT137831与2-脱氧-D-葡萄糖联用, 可以明显抑制葡萄糖和谷氨酰胺代谢以及肿瘤细胞在体内外生长。
白蔹素(ampelopsin)是一种黄烷醇, 主要存在于蛇葡萄属植物中。NOX4可在内质网中合成和折叠, 其在内质网应激中发挥重要作用。白蔹素能够明显升高细胞ROS水平, 产生内质网应激, 促进结肠细胞的凋亡, 但这一效应能够被NAC逆转, 提示白蔹素可能是通过ROS介导的机制产生上述效应[36]。此外白蔹素与EGFR抑制剂埃罗替尼联合应用可显著诱导EGFR突变的耐药NSCLC细胞NOX2表达上调, 使ROS水平升高, 最终导致caspase依赖性细胞死亡[37]。
辣椒素(capsaicin)是一类存在于辣椒中的天然产物, 是一种有效的NOX抑制剂[38]。有研究表明, 表达K-ras的人胰管上皮细胞E6E7中NOX高度活化和ROS水平较高。辣椒素能够抑制NOX的酶活性, 从而升高K-ras细胞中活性氧的水平, 导致选择性细胞增殖和侵袭性, 在AsPC-1细胞老鼠异种移植模型中具有抗肿瘤效果[39]。
3.3 靶向谷胱甘肽系统GSH由谷氨酸(Glu)、半胱氨酸(Cys)和甘氨酸(Gly)组成, 是生物体内一种重要的抗氧化剂, 能保护细胞膜中含巯基的蛋白质和酶不被氧化, 降低有毒外源性物质如自由基、过氧化物、脂质过氧化物和重金属对细胞元件的氧化损伤[40]。多种类型肿瘤组织及细胞内GSH浓度可原发或继发性的增高。肿瘤细胞内GSH浓度增加与烷化剂、多柔比星及铂类等药物耐药密切有关。肿瘤细胞中还原型GSH可与ROS作用转化为氧化型GSSG, 有效降低高浓度ROS导致的肿瘤细胞凋亡; 其次GSH上的巯基能够与具有亲电性片段的化合物共价结合, 降低某些抗肿瘤药物的细胞杀伤作用。在耐药肿瘤细胞中, GSH还作为辅助因子, 促进多药耐药蛋白-2 (multidrug resistance protein 2, MRP2)介导的某些抗癌药物外排。因此, 降低肿瘤GSH水平成为一种新型肿瘤治疗策略, 目前主要有直接消耗GSH, 或者调控GSH的产生与维持两种思路。根据药物的作用机制不同, 调控GSH系统的药物包括GSH清除剂、γ-谷氨酰半胱氨酸合成酶(γ-glutamylcysteine synthetase, γ-GCS)抑制剂、谷胱甘肽过氧化物酶(glutathione peroxidase, GPX)抑制剂、谷胱甘肽-S-转移酶(glutathione S-transferases, GSTs)抑制剂。
3.3.1 消耗细胞内GSHAPR-246是一个前体药物, 在体内可被生物转化为活性化合物亚甲基醌(MQ)[41], 与谷胱甘肽的半胱氨酸残基发生迈克尔加成形成共价结合, 消耗细胞内游离GSH。APR-246可以提高顺铂和多柔比星在p53突变的耐药卵巢肿瘤细胞中的抗肿瘤作用, 与吉西他滨或多西他赛联用也能产生明显的协同作用。APR-246通过消耗谷胱甘肽来提高对化疗药物的敏感性, 可以克服NCI-H2228 (Q331)细胞中缺氧诱导的顺铂耐药[42]。在NSCLC细胞系中, APR-246与多聚ADP-核糖聚合酶[poly(ADP-ribose) polymerase, PARP]抑制剂奥拉帕尼(AZD2281)表现出协同效应; 在AML细胞中, 与阿糖胞苷、柔红霉素、氟达拉滨发挥协同作用。目前APR-246以单独使用或联合用药的方式, 正在进行多项抗肿瘤治疗的临床试验(NCT03391050、NCT03268382、NCT02098343)。
异硫氰酸酯类化合物是一类广泛存在于十字花科植物中的天然产物, 其结构中含有异硫氰酸基团(-N=C=S), 可与亲电物质发生作用, 具有多重生物活性。其中异硫氰酸苯乙酯(phenethyl isothiocyanate, PEITC)是一种含有芳环的异硫氰酸酯类化合物, 在体内可与含有巯基的片段发生亲电加成反应, 消耗细胞GSH, 提高细胞ROS水平[43]。将PEITC分别与Bcr-Abl突变的白血病细胞系和来自CML患者的白血病细胞进行孵化, 可提高其细胞ROS水平, 促进Bcr-Abl蛋白降解, 导致白血病细胞凋亡, 而这一效应可被抗氧化剂N-乙酰半胱氨酸(N-acetycysteine, NAC)逆转。PEITC作为膳食补充剂正在开展头颈部肿瘤患者临床试验(NCT03034603)。另外萝卜籽中提取物莱菔素(SFE)同样具有异硫氰酸基团, 研究显示莱菔素通过耗竭GSH和诱导微管解聚抑制结肠癌实体瘤的生长[44]。图 5显示了部分调节GSH和γ-GCS的化合物。

|
Figure 5 GSH depletors |
γ-GCS是GSH生物合成中限速酶, 催化谷氨酸和半胱氨酸的结合, 是体内合成GSH的第一步, 对调节体内GSH的平衡发挥重要作用, 同时也能调节某些肿瘤对放化疗的敏感性。抑制γ-GCS的活性能够显著降低内源性GSH的合成, 是降低肿瘤细胞内GSH的有效途径[45]。γ-GCS抑制剂丁硫氨酸亚砜胺(buthionine sulfoximine, BSO) (图 6)能显著降低细胞GSH水平, 提高多种抗肿瘤药物的活性以及其细胞毒性, 包括逆转乳腺癌细胞中KLF-4介导的顺铂耐药[46]以及急性早幼粒细胞白血病细胞中三氧化二砷耐药[47]。目前BSO联合美法仑(melphalan)已经完成治疗难治性高危神经母细胞瘤的Ⅰ期临床试验(NCT00005835)[48]。

|
Figure 6 γ-GCS inhibitors |
GSTs是一种Ⅱ相代谢酶, 能催化还原GSH巯基与亲电性底物结合生成水溶性更高、极性更大的复合物, 这些复合物又可被多药耐药蛋白(MRP)和P-糖蛋白(P-gp)等泵出体外, 使机体解毒[49]。当GSTs表达上调或活性增强时, 药物外排将会增加, 导致其在体内作用时间变短, 不能有效发挥作用从而产生耐药性。根据氨基酸序列的相似性、基因序列以及免疫交叉反应性, 细胞质GSTs可分为7个亚型: α、μ、π、σ、θ、ω和ζ, 其中GSTπ与人类细胞的癌变、肿瘤的形成及肿瘤细胞耐药性的产生密切相关。GSTπ在大多数肿瘤细胞尤其是在化疗药物耐药的肿瘤细胞中都有很高的表达, 其被认为是肝癌形成的标志之一, 并且在肺癌、结肠癌、肾癌、卵巢癌、食道癌和胃癌中均发现了高表达的GSTπ[50, 51]。GSTπ-1过表达可降低HeLa细胞的放疗效果[52], 对胃癌预后也有负面影响[53]。通过siRNA抑制GSTπ表达, 能显著增加顺铂在耐药卵巢癌细胞内积累并恢复其化疗效果[54], 提示靶向GSTπ可能是潜在抗耐药肿瘤作用靶点。
依他尼酸(etacrynic acid, EA) (图 7)是一类常见的利尿剂, 临床上主要用于治疗各种原因引起的水肿。EA结构中具有α, β-不饱和酮, 可与GSH发生迈克尔加成反应形成共价结合消耗GSH, 同时也可抑制多种GSTs同工酶, 增加细胞对化疗药物的敏感性。此外EA通过烯基和GSH的巯基结合形成EA-GSH结合物(EA-GSH), 该化合物对GSTπ的抑制活性强于EA (KiEA-GSH = 11.5 μmol·L-1, KiEA = 1.5 μmol·L-1)[55]。

|
Figure 7 GST inhibitors |
6-(7-Nitro-2, 1, 3-benzoxadiazol-4-ylthio)hexanol (NBDHEX)是一类经典的GST抑制剂, 对GSTπ具有很高的亲和性[56]。NBDHEX可激活DOX耐药的小细胞肺癌H69AR细胞中caspase依赖性凋亡, 激活c-Jun激酶, 降低抗凋亡蛋白Bcl-2的表达[57]。NBDHEX对P-gp高表达的急性髓系白血病细胞也具有细胞毒性, 还能作为增敏剂增强替莫唑胺在A375黑色素瘤细胞中的抗增殖作用[58]。
3.3.4 靶向xCTxCT (又称谷氨酸/胱氨酸逆向转运蛋白溶质载体家族7号成员11, SLC7A11)属于逆向转运系统Xc-, 介导细胞外L-胱氨酸和细胞内L-谷氨酸在质膜上的交换, 为胞内GSH合成提供半胱氨酸[59]。敲除xCT则可降低GSH水平, 增加细胞本身ROS水平, 导致肿瘤细胞死亡增加[60]。xCT可参与多项生理过程, 如介导体细胞和免疫细胞的生存, 保护正常细胞免受致癌物的氧化损伤, 同时还与癌细胞的生长和恶性进展、对化疗的抵抗、促进肿瘤干细胞样表型化等相关。临床研究表明, xCT表达上调可促进舌鳞癌中的顺铂耐药和成胶质细胞瘤中替莫唑胺(TMZ)耐药[61]; xCT表达上调还是结肠直肠癌和食管鳞癌复发的独立预测因子[62]。以上研究表明xCT是一个潜在的抗肿瘤靶点, 常见的xCT抑制剂的结构如图 8所示。

|
Figure 8 xCT inhibitors |
柳氮磺胺吡啶(sulfasalazine)是一种磺胺类抗菌药, 临床上主要用于治疗类风湿性关节炎和炎症性肠病, 同时也是一类有效的xCT抑制剂[63, 64]。柳氮磺胺吡啶(100 μmol·L-1)能有效抑制CD133-阳性肝肿瘤细胞xCT活性, 削弱ROS防御系统, 增加顺铂和DOX在CD133-阳性肝癌细胞中增殖抑制作用[65]。另外, CD44是透明质酸的受体, 通常在胚胎干细胞、造血干细胞和癌症干细胞中表达, CD44v是细胞表面蛋白CD44的变体亚型, 它可稳定xCT并调节癌细胞中已降低的GSH胞内水平[66]。目前柳氮磺胺吡啶正在对难以用顺铂治疗的CD44v阳性胃癌患者进行临床试验[67], 用于预防接受盆腔放疗的癌症患者的急性腹泻。
Erastin是一种电压依赖性阴离子通道结合的小分子, 在抑制xCT方面比柳氮磺胺吡啶更有效。Erastin通过抑制xCT使肿瘤细胞GSH水平降低, 从而增加了替莫唑胺(TMZ)的细胞毒性[68]。此外, Erastin还可激活细胞铁死亡(ferroptosis)[69]。Erastin (5 μmol·L-1)可通过诱发铁死亡和坏死性凋亡, 杀死阿糖胞苷和DOX耐药的急性髓细胞性白血病细胞。Erastin (2.5或5 μmol·L-1)与大麻二酚联合使用可发挥协同作用, 通过增加肿瘤活性氧的水平, 抑制神经胶质瘤干细胞的生存、入侵和自我更新[70]。
3.4 靶向硫氧还蛋白还原酶硫氧还蛋白系统由硫氧还蛋白(Trx)、硫氧还蛋白还原酶(TrxR)和NADPH共同构成, 是调节细胞内氧化还原平衡的重要防御系统, 此外还涉及细胞内多种生物功能, 例如脱氧核苷酸合成、细胞生长、凋亡和细胞信号转导等[71]。其中TrxRs催化NADPH依赖的氧化态Trx还原, 以及其他内源性和外源性化合物的还原, 以调节细胞内氧化平衡。在肿瘤发展的不同阶段, Trx在癌变细胞中发挥着不同的功能。在肿瘤发展早期, Trx通过抵抗致癌物引起的氧化应激, 能够抑制肿瘤生成; 但当细胞开始出现癌症表型时, 高水平的Trx和TrxR能够促进细胞增长和抵抗细胞凋亡, 因此又可能会促进肿瘤的发展; 在肿瘤发展的稍后阶段, Trx还能够调节血管内皮生长因子和基质金属蛋白酶, 促进肿瘤血管的生成和转移[72]。TrxR在肿瘤中过度表达, 其表达水平高低与肿瘤细胞的恶性程度、增殖和转移能力以及耐药水平密切相关, 抑制TrxR可以作为肿瘤治疗以及克服肿瘤细胞耐药的一个新策略。TrxR抑制剂按其结构可分为含有金属离子以及金属配合物、含迈克尔受体的化合物、含有硒的化合物和硝基芳香化合物, 图 9显示了部分的TrxR抑制剂结构。

|
Figure 9 TrxR inhibitors |
金诺芬(auranofin, AF)是一种含有金原子的口服抗炎药, 临床用于治疗类风湿性关节炎。现有研究表明AF能有效抑制还原态TrxR, 诱导肿瘤细胞凋亡[73]。该药物目前正在进行治疗卵巢癌的Ⅱ期临床试验(NCT03456700)。在TrxR过表达和耐DOX白血病细胞(K562/ADM)中, AF能显著抑制细胞增殖和诱导凋亡[74]。AF (10 mg·kg-1)与DOX联合用药可对A549小鼠异种移植瘤模型产生协同抗肿瘤效应。
含有迈克尔受体的多酚类化合物姜黄素(curcumin)具有抗氧化、抗肿瘤和抗炎等多种生物活性和药理作用。姜黄素可以与TrxR蛋白C末端活性位点的Cys残基和Ser残基产生共价作用而不可逆地抑制其活性, 通过促氧化作用诱导肿瘤细胞死亡。在对姜黄素的结构改造中, 采用呋喃环替代姜黄素苯环合成了一系列姜黄素类似物, 这类化合物也通过靶向细胞内的TrxR而诱导细胞凋亡, 其中化合物2a对TrxR抑制作用达到纳摩尔级别[75]。化合物10是Ai等[76]合成的一种新型川芎嗪-姜黄素杂合物, 该结构中同样含有迈克尔受体结构, 能显著抑制对药敏感(A549、SPC-A-1和LTEP-G-2)和耐药(A549/DDP)肺癌细胞的增殖, 但对非肿瘤肺上皮样细胞无显著影响。化合物10可抑制TrxR/Trx系统和增加细胞内ROS积累和肿瘤凋亡。
依他塞伦(ethaselen)是一种有机硒类TrxR1抑制剂, 与某些化疗药物和放疗联合使用具有增敏作用。依他塞伦与顺铂联用, 可显著增加顺铂对耐药性人类红细胞白血病K562/CDDP细胞的增殖抑制作用[77]。依他塞伦还能通过类似的机制, 与舒尼替尼(多靶点酪氨酸激酶抑制剂)产生协同作用, 诱导大肠癌细胞的凋亡。还有其他含硒黄酮衍生物, 同样能够有效抑制TrxR, 提高耐药肿瘤细胞对药物的敏感性[78]。
3.5 靶向Nrf2Nrf2是体内重要的细胞保护性转录因子, 与Keap1和抗氧化反应元件(ARE)一起, 通过调节众多细胞保护性基因的表达来保护细胞。Keap1-Nrf2-ARE通路是细胞对ROS和亲电试剂引起的内源性和外源性氧化应激反应的主要调控途径。在非应激条件下, Keap1与Nrf2结合, 通过泛素化促进Nrf2降解。然而, 在氧化应激水平较高的环境中, 诱导物(如内源性或外源性ROS)修饰Keap1半胱氨酸残基, 通过解离Keap1-Nrf2复合物抑制Nrf2泛素化。然后Nrf2转移到细胞核并与ARE结合, 诱导抗氧化剂或解毒蛋白的表达[79, 80]。
Nrf2在肿瘤的发生和发展中表现出两面性。在肿瘤发生的早期, Nrf2的活化能够有效加速异物代谢和维持微环境稳态, 从而抑制肿瘤的发生。而在肿瘤发展和恶化阶段, Nrf2则表现出较明显癌蛋白性质。Nrf2的异常活化可激活其下游基因转录, 进而促进肿瘤的发生和发展[81]。Nrf2可下调肿瘤细胞内活性氧水平, 抑制依赖氧化应激的肿瘤化疗药物的活性; 上调谷胱甘肽含量, 促进抗肿瘤药物代谢; 促进药物外排泵表达, 降低细胞内药物浓度[82]。通过上述途径, Nrf2的活化可有效促进肿瘤细胞存活、增强其耐药性。同时Nrf2的代谢调控作用可促进肿瘤细胞代谢重编程:促进葡萄糖摄取并通过无氧酵解途径代谢; 增加核苷合成和还原当量NADPH的供应; 调节氨基酸代谢, 促进丝氨酸/甘氨酸合成。通过这些代谢途径改变, 使细胞内的能量和物质代谢能够满足肿瘤快速增殖的需求[79]。以上研究表明Nrf2是一个潜在的抗肿瘤靶点, 常见的Nrf2抑制剂的结构如图 10所示。

|
Figure 10 Nrf2 inhibitors |
化合物ML385是通过高通量筛选发现具有Nrf2-ARE抑制活性的化合物, 其能有效抑制Nrf2与靶基因的结合, 从而抑制其下游靶基因表达。ML385 (1或5 μmol·L-1)与铂类药物、DOX或紫杉醇联合使用时, 能够显著增加这些药物在NSCLC细胞中的细胞毒性。在Nrf2高度活化的NSCLC细胞中, ML385 (30 mg·kg-1)联合卡铂用药, 可产生显著抗肿瘤作用[83]。
4-(2-环己乙基)苯胺(IM3829)也是通过高通量筛选得到的活性化合物, 该化合物可抑制Nrf2的转录活性。IM3829联合放疗可显著降低肺癌H1299、A549、H460细胞的生存。此外, IM3829 (5 mg·kg-1)联合放疗对H1299或A549肺癌异种移植小鼠肿瘤生长的抑制作用强于单独放疗。
一些糖皮质激素也表现出强效的Nrf2抑制活性, 可在纳摩尔水平抑制Nrf活性, 进一步研究表明, 这种Nrf2抑制效应与糖皮质激素受体相关[84]。例如, 丙酸氯倍他索(clobetasol propionate)能够通过激活糖皮质激素受体, 促进Nrf2的泛素化降解, 进而抑制Nrf2活性[85]。
许多天然产物也被报道具有Nrf2抑制活性。Brusatol是较早被报道的一种强效Nrf2抑制剂[86]。Brusatol能有效降低细胞内的Nrf2水平, 增强包括A549细胞在内的多种肿瘤细胞对化疗药物的敏感性。随后的机制研究表明, brusatol的作用机制主要是通过抑制蛋白质翻译途径发挥作用。这种非选择性的作用机制限制该化合物的进一步研究[87]。
一些黄酮和类黄酮类化合物也被报道具有Nrf2抑制活性。黄酮类化合物木犀草素(luteolin)在mRNA和蛋白水平上均抑制Nrf2, 导致Nrf2与ARE结合减少, 最终引起GSH耗竭[88]。木犀草素抑制直肠癌SW620-OX细胞中Nrf2靶基因NQO1、HO-1和GSTα1/2的表达, 使GSH水平显著降低。木犀草素(5 μmol·L-1)还能够显著提高奥沙利铂、顺铂和DOX在耐药细胞株HCT116-OX和SW620-OX中的抗肿瘤作用[89]。另一类黄酮类化合物汉黄芩素(wogonin)可通过抑制Nrf2-ARE通路增加HepG2细胞和MCF-7/DOX细胞对化疗药物敏感性[90, 91]。值得注意的是, 黄酮类化合物对Nrf2活性的调节表现出双面效应。特别是在非恶性转化的细胞中, 其能表现出Nrf2激活效应。例如汉黄芩素能通过激活Nrf2, 阻止结肠炎向结肠癌的转换[92]; 木樨草素也能通过激活Nrf2表现出一定肝脏保护和神经保护效应[93]。
4 结语由于外界刺激、代谢活性提高以及线粒体功能紊乱, 与正常细胞相比, 肿瘤细胞呈现出更高的ROS水平。一方面提髙抗氧化能力, 降低ROS水平可阻止癌症的发生; 另一方面由于肿瘤细胞自身具有更高的氧化应激水平, 其对ROS的进一步上升更加敏感。由于ROS在癌症发展中的双重作用, 使得抗肿瘤药物研发中, 抗氧化和促氧化策略同时受到关注。抗氧化可以清除ROS预防细胞癌变, 而促氧化剂可以促进癌细胞中ROS的生成或者减弱抗氧化能力从而破坏肿瘤细胞的氧化平衡, 促进细胞凋亡。当细胞发展为前癌细胞时, 再使用抗氧化剂已不能阻止癌细胞扩散, 因此需使用凋亡诱导试剂阻止癌症发展。
但是基于ROS调控的抗肿瘤药物研发依然存在一些关键问题需要解决。首先大多是ROS调控剂自身抗肿瘤活性较弱, 在肿瘤治疗中常作为化疗增敏剂或者辅助用药使用; 其次ROS调控剂多为非靶标特异性药物, 在使用过程中存在不良反应较多的情况, 以上均限制其在临床上的进一步使用。未来在此领域的研究, 一方面要注重提高ROS调控剂的抗肿瘤活性或是与其他药物联用起到协同作用, 同时还需要更多关注ROS调控剂对靶点的选择性。
通过促进ROS的产生, 破坏肿瘤细胞氧化还原平衡, 正在成为一种新型的抗肿瘤策略, 以上调ROS为目标的化合物已经在开展抗肿瘤的临床研究。本综述对调节ROS水平的抗肿瘤药物进行总结(表 1), 希望能够为此领域的研究提供思路和参考。
| Table 1 Summary of structures of ROS modulators |

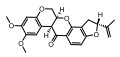
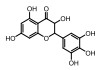
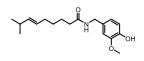
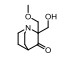
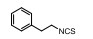
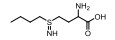

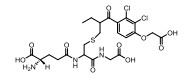


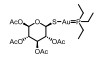

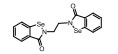
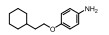
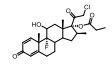
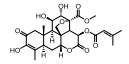
| [1] |
Del Rio LA, Sandalio LM, Palma JM, et al. Metabolism of oxygen radicals in peroxisomes and cellular implications[J]. Free Radic Biol Med, 1992, 13: 557-580. DOI:10.1016/0891-5849(92)90150-F |
| [2] |
Inoue M, Sato EF, Nishikawa M, et al. Mitochondrial generation of reactive oxygen species and its role in aerobic life[J]. Curr Med Chem, 2003, 10: 2495-2505. DOI:10.2174/0929867033456477 |
| [3] |
Kumari S, Badana AK, Mohan GM, et al. Reactive oxygen species: a key constituent in cancer survival[J]. Biomark Insights, 2018, 13: 1-9. |
| [4] |
Bridge G, Rashid S, Martin SA. DNA mismatch repair and oxidative DNA damage: implications for cancer biology and treatment[J]. Cancers (Basel), 2014, 6: 1597-1614. DOI:10.3390/cancers6031597 |
| [5] |
Liou GY, Storz P. Reactive oxygen species in cancer[J]. Free Radic Res, 2010, 44: 479-496. DOI:10.3109/10715761003667554 |
| [6] |
Hou GX, Liu PP, Zhang S, et al. Elimination of stem-like cancer cell side-population by auranofin through modulation of ROS and glycolysis[J]. Cell Death Dis, 2018, 9: 89. DOI:10.1038/s41419-017-0159-4 |
| [7] |
Wenzel P, Kossmann S, Munzel T, et al. Redox regulation of cardiovascular inflammation - immunomodulatory function of mitochondrial and Nox-derived reactive oxygen and nitrogen species[J]. Free Radic Biol Med, 2017, 109: 48-60. DOI:10.1016/j.freeradbiomed.2017.01.027 |
| [8] |
Mittal M, Siddiqui MR, Tran K, et al. Reactive oxygen species in inflammation and tissue injury[J]. Antioxid Redox Signal, 2014, 20: 1126-1167. DOI:10.1089/ars.2012.5149 |
| [9] |
Ahmad S, Khan H, Shahab U, et al. Protein oxidation: an overview of metabolism of sulphur containing amino acid, cysteine[J]. Front Biosci (Schol Ed), 2017, 9: 71-87. DOI:10.2741/s474 |
| [10] |
Stewart TM, Dunston TT, Woster PM, et al. Polyamine catabolism and oxidative damage[J]. J Biol Chem, 2018, 293: 18736-18745. DOI:10.1074/jbc.TM118.003337 |
| [11] |
Brewer TF, Garcia FJ, Onak CS, et al. Chemical approaches to discovery and study of sources and targets of hydrogen peroxide redox signaling through NADPH oxidase proteins[J]. Annu Rev Biochem, 2015, 84: 765-790. DOI:10.1146/annurev-biochem-060614-034018 |
| [12] |
Ma L, Fu Q, Xu B, et al. Breast cancer-associated mitochondrial DNA haplogroup promotes neoplastic growth via ROS-mediated AKT activation[J]. Int J Cancer, 2018, 142: 1786-1796. DOI:10.1002/ijc.31207 |
| [13] |
Cadet J, Wagner JR. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation[J]. Cold Spring Harb Perspect Biol, 2013, 5: a012559. |
| [14] |
Tretyakova NY, Groehler AT, Ji S. DNA-protein cross-links: formation, structural identities, and biological outcomes[J]. Acc Chem Res, 2015, 48: 1631-1644. DOI:10.1021/acs.accounts.5b00056 |
| [15] |
Brovkovych V, Aldrich A, Li N, et al. Removal of serum lipids and lipid-derived metabolites to investigate breast cancer cell biology[J]. Proteomics, 2019, 19: e1800370. |
| [16] |
Maiorino M, Conrad M, Ursini F. GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues[J]. Antioxid Redox Signal, 2018, 29: 61-74. DOI:10.1089/ars.2017.7115 |
| [17] |
Zafar A, Singh S, Satija YK, et al. Deciphering the molecular mechanism underlying anticancer activity of coumestrol in triple-negative breast cancer cells[J]. Toxicol In Vitro, 2018, 46: 19-28. DOI:10.1016/j.tiv.2017.10.007 |
| [18] |
Cui Q, Wang JQ, Assaraf YG, et al. Modulating ROS to overcome multidrug resistance in cancer[J]. Drug Resist Updat, 2018, 41: 1-25. DOI:10.1016/j.drup.2018.11.001 |
| [19] |
Birben E, Sahiner UM, Sackesen C, et al. Oxidative stress and antioxidant defense[J]. World Allergy Organ J, 2012, 5: 9-19. DOI:10.1097/WOX.0b013e3182439613 |
| [20] |
Parekh A, Das S, Parida S, et al. Multi-nucleated cells use ROS to induce breast cancer chemo-resistance in vitro and in vivo[J]. Oncogene, 2018, 37: 4546-4561. DOI:10.1038/s41388-018-0272-6 |
| [21] |
Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy[J]. Nat Chem Biol, 2015, 11: 9-15. DOI:10.1038/nchembio.1712 |
| [22] |
Jonckheere AI, Smeitink JA, Rodenburg RJ. Mitochondrial ATP synthase: architecture, function and pathology[J]. J Inherit Metab Dis, 2012, 35: 211-225. DOI:10.1007/s10545-011-9382-9 |
| [23] |
Jastroch M, Divakaruni AS, Mookerjee S, et al. Mitochondrial proton and electron leaks[J]. Essays Biochem, 2010, 47: 53-67. DOI:10.1042/bse0470053 |
| [24] |
Deribe YL, Sun Y, Terranova C, et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer[J]. Nat Med, 2018, 24: 1627. |
| [25] |
Petrosillo G, Ruggiero FM, Pistolese M, et al. Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis[J]. FEBS Lett, 2001, 509: 435-438. DOI:10.1016/S0014-5793(01)03206-9 |
| [26] |
Molina JR, Sun Y, Protopopova M, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability[J]. Nat Med, 2018, 24: 1036-1046. DOI:10.1038/s41591-018-0052-4 |
| [27] |
Rohlena J, Dong LF, Ralph SJ, et al. Anticancer drugs targeting the mitochondrial electron transport chain[J]. Antioxid Redox Signal, 2011, 15: 2951-2974. DOI:10.1089/ars.2011.3990 |
| [28] |
Cierlitza M, Chauvistre H, Bogeski I, et al. Mitochondrial oxidative stress as a novel therapeutic target to overcome intrinsic drug resistance in melanoma cell subpopulations[J]. Exp Dermatol, 2015, 24: 155-157. DOI:10.1111/exd.12613 |
| [29] |
Wu L, Zhao J, Cao K, et al. Oxidative phosphorylation activation is an important characteristic of DOX resistance in hepatocellular carcinoma cells[J]. Cell Commun Signal, 2018, 16: 6. DOI:10.1186/s12964-018-0217-2 |
| [30] |
Lu B, Gong X, Wang ZQ, et al. Shikonin induces glioma cell necroptosis in vitro by ROS overproduction and promoting RIP1/RIP3 necrosome formation[J]. Acta Pharmacol Sin, 2017, 38: 1543-1553. DOI:10.1038/aps.2017.112 |
| [31] |
Fath MA, Diers AR, Aykin-Burns N, et al. Mitochondrial electron transport chain blockers enhance 2-deoxy-D-glucose induced oxidative stress and cell killing in human colon carcinoma cells[J]. Cancer Biol Ther, 2009, 8: 1228-1236. DOI:10.4161/cbt.8.13.8631 |
| [32] |
Wang L, Zhang X, Cui G, et al. A novel agent exerts antitumor activity in breast cancer cells by targeting mitochondrial complex Ⅱ[J]. Oncotarget, 2016, 7: 32054-32064. DOI:10.18632/oncotarget.8410 |
| [33] |
Weng MS, Chang JH, Hung WY, et al. The interplay of reactive oxygen species and the epidermal growth factor receptor in tumor progression and drug resistance[J]. J Exp Clin Cancer Res, 2018, 37: 61. DOI:10.1186/s13046-018-0728-0 |
| [34] |
Marlein CR, Zaitseva L, Piddock RE, et al. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts[J]. Blood, 2017, 130: 1649-1660. DOI:10.1182/blood-2017-03-772939 |
| [35] |
Ameziane-El-Hassani R, Schlumberger M, Dupuy C. NADPH oxidases: new actors in thyroid cancer?[J]. Nat Rev Endocrinol, 2016, 12: 485-494. DOI:10.1038/nrendo.2016.64 |
| [36] |
Weyemi U, Redon CE, Parekh PR, et al. NADPH oxidases NOXs and DUOXs as putative targets for cancer therapy[J]. Anticancer Agents Med Chem, 2013, 13: 502-514. |
| [37] |
Green DE, Murphy TC, Kang BY, et al. The Nox4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation[J]. Am J Respir Cell Mol Biol, 2012, 47: 718-726. DOI:10.1165/rcmb.2011-0418OC |
| [38] |
Park GB, Jeong JY, Kim D. Ampelopsin-induced reactive oxygen species enhance the apoptosis of colon cancer cells by activating endoplasmic reticulum stress-mediated AMPK/MAPK/XAF1 signaling[J]. Oncol Lett, 2017, 14: 7947-7956. |
| [39] |
Hong SW, Park NS, Noh MH, et al. Combination treatment with erlotinib and ampelopsin overcomes erlotinib resistance in NSCLC cells via the Nox2-ROS-Bim pathway[J]. Lung Cancer, 2017, 106: 115-124. DOI:10.1016/j.lungcan.2017.02.009 |
| [40] |
Lin MH, Lee YH, Cheng HL, et al. Capsaicin inhibits multiple bladder cancer cell phenotypes by inhibiting tumor-associated NADH oxidase (tNOX) and sirtuin1 (SIRT1)[J]. Molecules, 2016, 21: e849. DOI:10.3390/molecules21070849 |
| [41] |
Wang P, Sun YC, Lu WH, et al. Selective killing of K-ras-transformed pancreatic cancer cells by targeting NAD(P)H oxidase[J]. Chin J Cancer, 2015, 34: 166-176. |
| [42] |
Bansal A, Simon MC. Glutathione metabolism in cancer progression and treatment resistance[J]. J Cell Biol, 2018, 217: 2291-2298. DOI:10.1083/jcb.201804161 |
| [43] |
Bykov VJ, Wiman KG. Mutant p53 reactivation by small molecules makes its way to the clinic[J]. FEBS Lett, 2014, 588: 2622-2627. DOI:10.1016/j.febslet.2014.04.017 |
| [44] |
Deben C, Deschoolmeester V, De Waele J, et al. Hypoxia-induced cisplatin resistance in non-small cell lung cancer cells is mediated by HIF-1 alpha and mutant p53 and can be overcome by induction of oxidative stress[J]. Cancers (Basel), 2018, 10: e126. DOI:10.3390/cancers10040126 |
| [45] |
Abbaoui B, Lucas CR, Riedl KM, et al. Cruciferous vegetables, isothiocyanates, and bladder cancer prevention[J]. Mol Nutr Food Res, 2018, 62: e1800079. DOI:10.1002/mnfr.201800079 |
| [46] |
Byun S, Shin SH, Park J, et al. Sulforaphene suppresses growth of colon cancer-derived tumors via induction of glutathione depletion and microtubule depolymerization[J]. Mol Nutr Food Res, 2016, 60: 1068-1078. DOI:10.1002/mnfr.201501011 |
| [47] |
Lin LC, Chen CF, Ho CT, et al. gamma-Glutamylcysteine synthetase (gamma-GCS) as a target for overcoming chemo- and radio-resistance of human hepatocellular carcinoma cells[J]. Life Sci, 2018, 198: 25-31. DOI:10.1016/j.lfs.2018.02.015 |
| [48] |
Jia Y, Zhang C, Zhou L, et al. Micheliolide overcomes KLF4-mediated cisplatin resistance in breast cancer cells by downregulating glutathione[J]. Onco Targets Ther, 2015, 8: 2319-2327. |
| [49] |
Davison K, Cote S, Mader S, et al. Glutathione depletion overcomes resistance to arsenic trioxide in arsenic-resistant cell lines[J]. Leukemia, 2003, 17: 931-940. DOI:10.1038/sj.leu.2402876 |
| [50] |
Villablanca JG, Volchenboum SL, Cho H, et al. A phase I new approaches to neuroblastoma therapy study of buthionine sulfoximine and melphalan with autologous stem cells for recurrent/refractory high-risk neuroblastoma[J]. Pediatr Blood Cancer, 2016, 63: 1349-1356. DOI:10.1002/pbc.25994 |
| [51] |
Chatterjee A, Gupta S. The multifaceted role of glutathione S-transferases in cancer[J]. Cancer Lett, 2018, 433: 33-42. DOI:10.1016/j.canlet.2018.06.028 |
| [52] |
O'brien ML, Tew KD. Glutathione and related enzymes in multidrug resistance[J]. Eur J Cancer, 1996, 32A: 967-978. |
| [53] |
Yang L, Liu R, Ma HB, et al. Radiosensitivity in HeLa cervical cancer cells overexpressing glutathione S-transferase pi 1[J]. Oncol Lett, 2015, 10: 1473-1476. DOI:10.3892/ol.2015.3394 |
| [54] |
Yu P, Cheng X, Du Y, et al. Significance of MDR-related proteins in the postoperative individualized chemotherapy of gastric cancer[J]. J Cancer Res Ther, 2015, 11: 46-50. DOI:10.4103/0973-1482.147695 |
| [55] |
Zhang T, Guan M, Jin HY, et al. Reversal of multidrug resistance by small interfering double-stranded RNAs in ovarian cancer cells[J]. Gynecol Oncol, 2005, 97: 501-507. DOI:10.1016/j.ygyno.2005.01.027 |
| [56] |
Ploemen JHTM, Vanommen B, Bogaards JJP, et al. Ethacrynic-acid and its glutathione conjugate as inhibitors of glutathione S-transferases[J]. Xenobiotica, 1993, 23: 913-923. DOI:10.3109/00498259309059418 |
| [57] |
Federici L, Lo Sterzo C, Pezzola S, et al. Structural basis for the binding of the anticancer compound 6-(7-nitro-2, 1, 3-benzoxadiazol-4-ylthio)hexanol to human glutathione S-transferases[J]. Cancer Res, 2009, 69: 8025-8034. DOI:10.1158/0008-5472.CAN-09-1314 |
| [58] |
Filomeni G, Turella P, Dupuis ML, et al. 6-(7-Nitro-2, 1, 3-benzoxadiazol-4-ylthio) hexanol, a specific glutathione S-transferase inhibitor, overcomes the multidrug resistance (MDR)-associated protein 1-mediated MDR in small cell lung cancer[J]. Mol Cancer Ther, 2008, 7: 371-379. DOI:10.1158/1535-7163.MCT-07-0487 |
| [59] |
De Luca A, Rotili D, Carpanese D, et al. A novel orally active water-soluble inhibitor of human glutathione transferase exerts a potent and selective antitumor activity against human melanoma xenografts[J]. Oncotarget, 2015, 6: 4126-4143. DOI:10.18632/oncotarget.2798 |
| [60] |
Arensman MD, Yang XS, Leahy DM, et al. Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity[J]. Proc Natl Acad Sci U S A, 2019, 116: 9533-9542. DOI:10.1073/pnas.1814932116 |
| [61] |
Ungard RG, Linher-Melville K, Nashed M, et al. xCT knockdown in human breast cancer cells delays onset of cancer-induced bone pain[J]. Mol Pain, 2019, 15: 1-14. |
| [62] |
Polewski MD, Reveron-Thornton RF, Cherryholmes GA, et al. Increased expression of system xc- in glioblastoma confers an altered metabolic state and temozolomide resistance[J]. Mol Cancer Res, 2016, 14: 1229-1242. DOI:10.1158/1541-7786.MCR-16-0028 |
| [63] |
Sugano K, Maeda K, Ohtani H, et al. Expression of xCT as a predictor of disease recurrence in patients with colorectal cancer[J]. Anticancer Res, 2015, 35: 677-682. |
| [64] |
Nunokawa T, Yokogawa N, Shimada K, et al. Effect of sulfasalazine use on the presence of Pneumocystis organisms in the lung among patients with rheumatoid arthritis: a test-negative design case-control study with PCR tests[J]. Mod Rheumatol, 2019, 29: 436-440. DOI:10.1080/14397595.2018.1465647 |
| [65] |
Wada F, Koga H, Akiba J, et al. High expression of CD44v9 and xCT in chemoresistant hepatocellular carcinoma: potential targets by sulfasalazine[J]. Cancer Sci, 2018, 109: 2801-2810. DOI:10.1111/cas.13728 |
| [66] |
Ogihara K, Kikuchi E, Okazaki S, et al. Sulfasalazine could modulate the CD44v9-xCT system and enhance cisplatin-induced cytotoxic effects in metastatic bladder cancer[J]. Cancer Sci, 2019, 110: 1431-1441. DOI:10.1111/cas.13960 |
| [67] |
Shitara K, Doi T, Nagano O, et al. Phase 1 study of sulfasalazine and cisplatin for patients with CD44v-positive gastric cancer refractory to cisplatin (EPOC1407)[J]. Gastric Cancer, 2017, 20: 1004-1009. DOI:10.1007/s10120-017-0720-y |
| [68] |
Chen L, Li X, Liu L, et al. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-gamma-lyase function[J]. Oncol Rep, 2015, 33: 1465-1474. DOI:10.3892/or.2015.3712 |
| [69] |
Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death[J]. Cell, 2012, 149: 1060-1072. DOI:10.1016/j.cell.2012.03.042 |
| [70] |
Singer E, Judkins J, Salomonis N, et al. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma[J]. Cell Death Dis, 2015, 6: e1601. DOI:10.1038/cddis.2014.566 |
| [71] |
Zhang J, Li X, Han X, et al. Targeting the thioredoxin system for cancer therapy[J]. Trends Pharmacol Sci, 2017, 38: 794-808. DOI:10.1016/j.tips.2017.06.001 |
| [72] |
Bian M, Fan R, Zhao S, et al. Targeting the thioredoxin system as a strategy for cancer therapy[J]. J Med Chem, 2019, 62: 7309-7321. |
| [73] |
Chen X, Shi X, Wang X, et al. Novel use of old drug: anti-rheumatic agent auranofin overcomes imatinib-resistance of chronic myeloid leukemia cells[J]. Cancer Cell Microenviron, 2014, 1: e415. |
| [74] |
Liu JJ, Liu Q, Wei HL, et al. Inhibition of thioredoxin reductase by auranofin induces apoptosis in adriamycin-resistant human K562 chronic myeloid leukemia cells[J]. Pharmazie, 2011, 66: 440-444. |
| [75] |
Qiu X, Liu Z, Shao WY, et al. Synthesis and evaluation of curcumin analogues as potential thioredoxin reductase inhibitors[J]. Bioorg Med Chem, 2008, 16: 8035-8041. DOI:10.1016/j.bmc.2008.07.054 |
| [76] |
Ai Y, Zhu B, Ren C, et al. Discovery of new monocarbonyl ligustrazine-curcumin hybrids for intervention of drug-sensitive and drug-resistant lung cancer[J]. J Med Chem, 2016, 59: 1747-1760. |
| [77] |
Ye SF, Yang Y, Wu L, et al. Ethaselen: a novel organoselenium anticancer agent targeting thioredoxin reductase 1 reverses cisplatin resistance in drug-resistant K562 cells by inducing apoptosis[J]. J Zhejiang Univ Sci B, 2017, 18: 373-382. |
| [78] |
Zheng X, Zhang Y, Zhang L, et al. Synergistic inhibition of sunitinib and ethaselen against human colorectal cancer cells proliferation[J]. Biomed Pharmacother, 2016, 83: 212-220. DOI:10.1016/j.biopha.2016.06.040 |
| [79] |
Jiang ZY, Lu MC, You QD. Nuclear factor erythroid 2-related factor 2 (Nrf2) inhibition: an emerging strategy in cancer therapy[J]. J Med Chem, 2019, 62: 3840-3856. |
| [80] |
Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway[J]. Arch Toxicol, 2011, 85: 241-272. DOI:10.1007/s00204-011-0674-5 |
| [81] |
Chartoumpekis DV, Wakabayashi N, Kensler TW. Keap1/Nrf2 pathway in the frontiers of cancer and non-cancer cell metabolism[J]. Biochem Soc Trans, 2015, 43: 639-644. DOI:10.1042/BST20150049 |
| [82] |
Hayes JD, Mcmahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer[J]. Trends Biochem Sci, 2009, 34: 176-188. DOI:10.1016/j.tibs.2008.12.008 |
| [83] |
Singh A, Venkannagari S, Oh KH, et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors[J]. ACS Chem Biol, 2016, 11: 3214-3225. DOI:10.1021/acschembio.6b00651 |
| [84] |
Kratschmar DV, Calabrese D, Walsh J, et al. Suppression of the Nrf2-dependent antioxidant response by glucocorticoids and 11beta-HSD1-mediated glucocorticoid activation in hepatic cells[J]. PLoS One, 2012, 7: e36774. DOI:10.1371/journal.pone.0036774 |
| [85] |
Choi EJ, Jung BJ, Lee SH, et al. A clinical drug library screen identifies clobetasol propionate as an NRF2 inhibitor with potential therapeutic efficacy in KEAP1 mutant lung cancer[J]. Oncogene, 2017, 36: 5285-5295. DOI:10.1038/onc.2017.153 |
| [86] |
Ren D, Villeneuve NF, Jiang T, et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism[J]. Proc Natl Acad Sci U S A, 2011, 108: 1433-1438. DOI:10.1073/pnas.1014275108 |
| [87] |
Vartanian S, Ma TP, Lee J, et al. Application of mass spectrometry profiling to establish brusatol as an inhibitor of global protein synthesis[J]. Mol Cell Proteomics, 2016, 15: 1220-1231. DOI:10.1074/mcp.M115.055509 |
| [88] |
Tang X, Wang H, Fan L, et al. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs[J]. Free Radic Biol Med, 2011, 50: 1599-1609. DOI:10.1016/j.freeradbiomed.2011.03.008 |
| [89] |
Chian S, Li YY, Wang XJ, et al. Luteolin sensitizes two oxaliplatin-resistant colorectal cancer cell lines to chemotherapeutic drugs via inhibition of the Nrf2 pathway[J]. Asian Pac J Cancer Prev, 2014, 15: 2911-2916. DOI:10.7314/APJCP.2014.15.6.2911 |
| [90] |
Qian C, Wang Y, Zhong Y, et al. Wogonin-enhanced reactive oxygen species-induced apoptosis and potentiated cytotoxic effects of chemotherapeutic agents by suppression Nrf2-mediated signaling in HepG2 cells[J]. Free Radic Res, 2014, 48: 607-621. DOI:10.3109/10715762.2014.897342 |
| [91] |
Zhong Y, Zhang F, Sun Z, et al. Drug resistance associates with activation of Nrf2 in MCF-7/DOX cells, and wogonin reverses it by down-regulating Nrf2-mediated cellular defense response[J]. Mol Carcinog, 2013, 52: 824-834. DOI:10.1002/mc.21921 |
| [92] |
Yao J, Zhao L, Zhao Q, et al. NF-kappaB and Nrf2 signaling pathways contribute to wogonin-mediated inhibition of inflammation-associated colorectal carcinogenesis[J]. Cell Death Dis, 2014, 5: e1283. DOI:10.1038/cddis.2014.221 |
| [93] |
Xu J, Wang H, Ding K, et al. Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE pathway[J]. Free Radic Biol Med, 2014, 71: 186-195. |