 2020, Vol. 55
2020, Vol. 55



药物的靶向和脱靶分别产生相应的药理作用和不良反应, 根源是药物分子与相应的生物大分子的微观结构互补性结合所致。药物在体内的药代动力学(吸收、分布、代谢、排泄等)和生物药剂学行为(溶解性、分配性、稳定性和荷电性等)主要由分子的宏观性质所决定, 微观结构和宏观性质都寓于化学结构之中。由先导物演化成候选药物的结构优化过程, 是将药理活性(强度和选择性)和成药性(药代、安全性和物化性质)以最佳配置融合于结构之中。药物化学的原理和内涵实质上是调整二者的兼顾性。
药物中有一类称作大环小分子药物, 分子中含有大约12~20 (或更多)个原子构成的大环结构。临床已有的大环药物有68个, 大都是环肽和大环内酯, 分别作注射和口服用药, 主要用于感染、炎性和肿瘤病上。线性肽环合成环肽, 可增加肽的稳定性和活性, 多是人为设计; 而大环内酯多为天然产物或其改构物, 较少分子设计所得。
近年来研究大环小分子药物成为新药研究的活跃领域, 动因是传统小分子对一些靶标(或靶标的突变)成药性空间窄促, 需要加以扩展, 熟知的类药5原则(RO5)束缚了人们研发设计的空间, 大环化合物是一种策略。
基于受体结构研制小分子药物中, 若在化学结构的适宜部位连接成大环, 对提高活性和选择性、药代和稳定性有重要影响。大环的形成是在优化结构的某个阶段, 将柔性的开链分子连接成有一定刚性的环状分子, 从而把线型分子的多种构象固定为宜于同靶标结合的活性构象, 降低了结合能的熵损失, 从而可提高活性和选择性。分子成环状结构往往也提高过膜和吸收性以及代谢稳定性, 所以, 研制大环小分子药物兼具提高活性和成药性的双重含义[1]。本文从上市或进入临床研究的大环药物的结构演化过程, 讨论研制大环小分子药物的策略理念。
1 结构生物学引导的大环设计当今大环小分子药物研发模式, 仍是按照常规的药物化学, 优化苗头或先导物, 基于化合物与靶标的共晶结构进行理性设计, 经构效关系(sar)和试错试验(trial and error)演化成高质量的活性分子, 在成环的过程中, 仍是在共晶结构指引下, 辅以动力学模拟和/或量子化学计算, 对线型活性分子作环合操作, 通过环合位点、尺寸大小和元素构成的探索性设计, 在sar和试错试验中得到优化的大环结构。大环的形成多是在优化的中后期完成。
1.1 从开链的克唑替尼到大环劳拉替尼辉瑞公司于2011年研发上市的克唑替尼(1, crizotinib), 针对的靶标是间变性淋巴瘤激酶(anaplastic lymphoma kinase, ALK)。ALK与核磷蛋白(nucleophosmin, NPM)的融合蛋白是发生间变性淋巴瘤和非小细胞肺癌(NSCLC)的关键性酶。然而使用1过程中出现耐药性和难以进入中枢的缺点(NSCLC常常发生脑转移), 为了克服耐药性和能够透过血脑屏障, 辉瑞研制出新一代ALK抑制剂劳拉替尼(2, lorlatinib), 为大环药物, 于2018年上市。
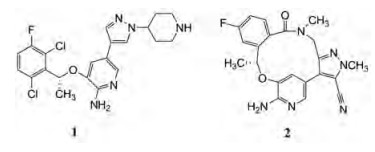
|
结构生物学提示, 耐受克唑替尼患者的ALK激酶显示氨基酸残基的多位点突变或插入, 例如L1196M、G1269A和G1202R, 这些氨基酸是与克唑替尼接触的残基, L1196作为门户残基, 是最容易发生变异的位点。
克唑替尼与ALK野生型和L1196M突变型的共晶结构表明, 分子的定位取向和结合模式几乎完全相同。仔细分析比较ALK空蛋白(apo)与ALK-克唑替尼复合物的晶体结构, 发现分子中二氯氟苯基的2-位氯原子与G1269骨架的羰基过于靠近(3.0 Å), 位阻效应迫使羰基扭转了30°, 因而支付了构象熵的耗能, 预示2-氯对结合是不利的因素, 如图 1所示。

|
Figure 1 Co-crystal structure of crizotinib with ALK wt (gray) overlaid with L1196M mutant (cyan) and the G1269 residue from L1196M apo structure |
图 2是以克唑替尼为起始物, 演化成劳拉替尼过程中的代表性化合物节点。去除2-Cl的化合物(1b)对发生L1196M突变的酶和细胞活性强于1a, 证实了共晶结构的分析。苯环的2位取代基和吡唑环分别加以变换, 1c显示有活性, 但低于1d, 1c的共晶结构显示(图 3, 青色分子), 酰胺的羰基氧经水分子的桥连, 与His 1124的骨架羰基形成氢键, 但不能与Lys1150发生氢键结合, 因为酰胺平面与苯环两面角呈45°, 此处的氢键形成需由近于垂直的两面角取向, 这可从图 3中1d (绿色分子)的2-位三唑环与苯环有的较大的两面角64°看出。因而1c氮上的甲基不能与G-环套上Leu1122、Gly1123和Val1130构成的疏水腔发生疏水-疏水相互作用, 这是1d活性强于1c的重要因素。同时, 晶体结构还显示了这些抑制剂结合态都呈U形构象, 提示头端的氟苯基与尾端的杂环在空间是接近的, 这成为进一步分子设计的切入点。

|
Figure 2 Evolutional scheme of crizotinib to macromolecule larlatinib through typical compounds |

|
Figure 3 ALK cocrystal of 1c (cyan) overlaid with 1d (green) showing the triazole, amide, and G-loop conformations |
开链分子的低能构象为了结合折叠“扭成”U形活性构象耗费了能量, 损失的构象熵可以通过连接首尾端基呈大环分子固定成U形构象加以消除, 同时连接的片段还可对疏水结合做出贡献, 为此需要能在此处同靶标发生疏水结合。成环后分子中的单键转动也受到限制, 从而可弥补简化结构损失的结合能, 也有利于穿越血脑屏障进入脑中。为此合成了1e, 活性非常强, 然而选择性不强。
分析ALK激酶的ATP结合腔, 是由27个氨基酸残基构成, 为了提高结合的选择性, 需针对与其他激酶ATP腔的不同组成上。在全部激酶家族中ALK激酶的Leu1198占26%, 而多数激酶(60%)为Phe或Tyr, Leu形状和体积不同于Phe和Tyr。抑制剂若只同Leu结合而与Phe和Tyr产生位阻, 应可以提高选择性。由于原肌球蛋白激酶(TrkB)相应于ALK Leu1198为Tyr, 因而TrkB作为评价大环的选择性活性的模型(抑制作用越弱越好)。化合物1e与ALK和TrkB的共晶图叠合表明, 吡唑环上甲基碳原子与TrkBTyr635的酚羟基(OH)、与羟基相连的碳(CZ)和邻位碳(CE)的距离都在原子的范德华半径范围内(图 4a和4b), 与Tyr635则无空间障碍, 可发生结合, 这是不利的, 实验表明1e对ALK缺乏选择性作用。在吡唑环上作不同的取代, SAR提示引入氰基的化合物(2)活性和选择性都很高, 图 4b是1e (绿色)和2 (青色)以及环丙基(黄色, 未编号)同ALK和TrkB的共晶叠合图, 氰基与环丙基同Tyr635的距离在原子的范德华半径之内, 发生不利的位阻效应, 实验结果确实提高了选择性。2通过血脑屏障的能力强于环丙基化合物, 从而确定为候选物, 研发成劳拉替尼[2]。

|
Figure 4 a. Compound 1e-ALK-bound structure (green) and 1e-TrkB (purple) with selectivity residues highlighted. PDB atom names for the terminal atoms are shown on Tyr635 (OH, CZ, CE). b. Ligands from ALK cocrystal structures aligned with TrkB, demonstrating the potential for clash between ligand and Tyr635 [1e (CH3, green), c-Pr (yellow), 2 (CN, cyan)]. Distances shown are between the 32 methyl carbon (shown as a ball) and TrkB Tyr635 |
表皮生长因子受体(EGFR)若缺失Ile19 (deI19)或突变Leu858为Arg (L858R), 则导致EGFR不依赖于配体的异常活化, 致使肿瘤细胞增殖, 特别是肺癌的发生。虽然已有多种EGFR激酶抑制剂(可逆和不可逆抑制)治疗非小细胞肺癌, 但患者用药大约两年后肺癌复发, 是由于EGFR另处发生T790M和C797S的变异而耐药, 导致所用药物失效。
勃林格因格翰公司研制的新一代抑制剂BI-4020是针对上述EGFRdeI19/T790M/C797S变异的激酶选择性抑制剂, 而对野生型EGFR无作用, 避免脱靶引起的不良反应。BI-4020为大环化合物, 临床试验效果良好。
为了寻找有初步选择性的先导化合物, 研制者用三突变的EGFR和野生型EGFR (WT)蛋白对化合物进行平行筛选评价, 发现3对EGFRL858R/T790M/C979S和对EGFRdeI19/T790M/C979S有一定活性, IC50分别为2 100和250 nmol·L-1, 而对野生型EGFR和其他激酶未见抑制作用。
3结合于EGFRL858R T790M的ATP结合腔(图 5), 共晶结构提示, 苯并咪唑环上N-H和羰基与EGFR铰链上的Met793结合, 羟异丁基占据核糖位置, 末端吡啶氮原子与Lys745形成氢键, 吡啶与苯环夹角40°, 为稳定构象(迫位无取代基的联芳基两面角40°为低能构象), 3与门户氨基酸Met790无空间障碍。
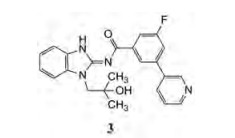
|

|
Figure 5 X-ray structure of ligand 3 bound to the ATP pocket of the EGFRL858R/T790M |
去掉3的苯环F原子, 酶学和细胞活性并未受到影响, 为了合成便捷, 后续优化在该位置不作取代。化合物3与EGFRL858R/T790M的共晶还提示如果将苯环换作吡啶(3a), 吡啶环N作为氢键接受体可与T854的羟基形成氢键, 而且不影响末端吡啶与Lys745的氢键结合。然而此时联吡啶上两个N在同侧, 形成偶极-偶极相互作用, 导致能量上不利, 通过量子力学-扭转角扫描也证实此时两个吡啶环呈大约30°的二面角, 并非能量上最低的160°。体外实验表明3a的活性强于3大约3~8倍(IC50 EGFRL858R/T790M/C797S= 353 nmol·L-1; IC50 EGFRdeI19/T790M/C797S = 57 nmol·L-1), 这与新增加的氢键弥补能量上损失有关。
化合物3b是末端换成了苯基(是去氟-3的区域异构体), 活性与3a相同, 而选择性更高, 虽然失去了末端吡啶N的氢键结合, 但也减少了偶极作用的斥力。共晶的吡啶-苯环的两面角为18°。而且与Lys745和Glu762的距离为3.5 Å, 避免了碰撞(图 6)。

|
Figure 6 X-ray structure of compound 3b |
基于共晶中3b的构象, 并且通过对3b构象的动力学分析和二维核磁的证实, 确定了末端苯环与羟异丁基靠近是活性构象, 如图 7所示。

|
Figure 7 Two-dimensional representation of the most prevalent conformations of 3b in complex with EGFRL858R T790M |
为此合成了大环分子3c, 对EGFRdel19 T790M C979S酶和相应的细胞活性分别高于3b大约17倍和5倍, 并且保持对野生型的低活性。3c的共晶(图 8)显示没有增加新的结合因素, 说明活性的提高是构象限制降低了能量的损失。共晶的重要启示是联吡啶-苯环的两面角41°, 与3a共晶的夹角相近, 说明末端芳环若有可与Lys745发生氢键结合的原子, 可增强与激酶的结合力。因而改变成N-甲基吡唑, 因为曾合成的开链吡唑环的活性与3a相近, 而代谢稳定性强于联吡啶化合物, 还考虑到吡啶的2位氟代可结合于该处的小腔, 故而合成了化合物3d, 对三突变的EGFR酶和细胞的抑制活性分别强于3c大约3倍和5倍。但3d的选择性低于3c, 为此将2-F变换成2-CH3 (3e), 3e对三突变的FGFR的酶和细胞活性维持不变, 选择性显著提高。此外, 考虑到这类大环的构象不是平坦的, 呈碗状构象, 有两种构象, 该不对称性可在大环的某处引入手性因素而加以固定, 以提高活性。

|
Figure 8 X-ray structure of compound 3c in complex with EGFRL85R T790M |
经分子模拟, 提示引入R-烷基可增强结合力, 合成的3f对EGFRdeI19 T790M C979S三突变的酶和细胞的活性显著提高, 而且也提高了选择性。3f的脂溶性强, 与血浆蛋白结合力过高, 从而将助溶基团连接在苯并咪唑的水相暴露区, 经过多次结构优化最终得到了化合物4, 代号为BI-4020, 其抗耐药性(三重突变的EGFR酶和依赖性细胞)、过膜性、溶解性和代谢稳定性都达到了既定的目标, 现处于临床研究阶段。图 9是用代表性的分子节点表示由开链的先导物3演化为大环候选药物BI-4020 (4)的示意图[3]。

|
Figure 9 The evolutional scheme of macromolecule BI-4020 (4) from open-chain molecule 3 |
凝血是个生化级联反应, 其中一个酶促环节是FXIa因子催化, 是胰蛋白酶样的丝氨酸蛋白酶, 在凝血酶生成的放大过程起关键作用, 因而对血栓的发生和成熟至关重要。血液中高水平的FXIa有发生心肌梗死和深静脉血栓的危险, 而FXIa低表达引起出血倾向较小, 因此, FXIa抑制剂应是出血风险低的抗血栓药物。
BMS公司研究咪唑类FXIa抑制剂, 经过SAR优化得到活性较好的开链分子5, 对FXIa的结合常数Ki = 5.8 nmol·L-1, 离体部分促凝血时间的活性aPTTEC1.5x = 5.3 μmol·L-1。为了进一步优化活性、药代和物化性质, 研究者采用大环分子策略, 开始于5与FXIa共晶结构设计的。
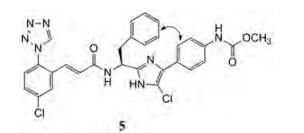
|
图 10的共晶结构显示, 5的高活性是由于多个杂原子参与了同XIa酶活性中心的结合, 表现在用虚线所示的氢键结合。P1和P2的苯环空间相隔较近, 确定为形成大环的位点, 如图 10中双箭头所示。过程如下:

|
Figure 10 X-ray crystal structure of acyclic phenyl imidazole 5 in factor XIa. The red spheres depict water molecules, and the dotted lines depict hydrogen bonds. The red arrows depict the potential for macrocyclization between the P1 prime and P2 prime groups |
① 用亚烷基或氧亚烷基(X=O)连接成12和13元环(n = 1, 2), 咪唑环的R可为H或Cl。然而所有合成的化合物活性都低于开链的先导物5, 活性最强的(n = 1, 饱和环, X=CH2, R=Cl)低于56倍。这些结果提示, 这些12/13元环的烷基或含醚键的烷基形成的大环构象与酶的契合达不到5的结合水平。另一高活性13元环化合物(n = 2, 饱和环, X=CH2, R=Cl)与XIa的共晶结构及其与5的结合构象作叠合提示, 参与大环形成的苯环距离酶的Leu41羰基过于靠近(3.2 Å), 位阻作用使苯环由原来开链的苯-咪唑的两面角25°扭转到53°, 这28°的变化虽然没有影响胺基甲酸酯的氢键结合, 但减弱了大环与S1处疏水腔的疏水相互作用。图 11a是13元环与酶的共晶结构图, 11b是13元环与5的活性构象的叠合图。
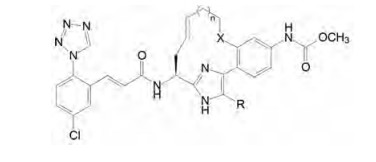
|

|
Figure 11 a. Overlay of X-ray crystal structures of acyclic phenyl imidazole 5 (yellow) and 13-membered macrocycle (cyan) in factor XIa. b. Overlay of X-ray crystal structures of acyclic phenyl imidazole 5 (yellow) and 13-membered macrocycle (cyan) in factor XIa looking into the S2 prime pocket. The increased dihedral angle in macrocycle pulled the macrocyclic linker out of the S1 prime pocket |
② 上述的13元环的苯基靠近Leu41提供了发生氢键结合的位点, 在大环与苯环的结点处引入酰胺片段, 发现酰胺型13元环的抑酶活性和抗凝血活性都强于5, 是因为增加了氢键结合的缘故。在咪唑环上引入氯原子活性提高10倍。
③ 在大环中引入双键, 反式体(6, E-form)的抑酶活性和抗凝血作用显著提高; 咪唑环引入氯原子, 化合物7Ki值达到最小, 不过抗凝活性未见提高。
化合物6与XIa的共晶结构提示大环中的CONH与Leu41发生氢键结合, 环中的反式双键使在S1'域的大环构象在适宜的空间, 没有与Cys42-Cys58的二硫键发生位阻作用(图 12)。6的口服生物利用度较低, 结构仍需优化[4]。

|
Figure 12 X-ray crystal structure of 13-membered unsaturated amide linked macrocycle 6 bound to factor XIa |
这是一个涉及大环、共价结合和消除寡肽骨架的分子操作, 体现了当今药物化学的研究内涵。研究的项目是近几十年来停滞不前的抗革兰阴性菌药物。
抗革兰阴性菌药物很少, 研制的难度在于同革兰阳性菌不同, 阴性菌有两层膜(内膜与外膜), 外膜上有脂多糖, 阻止药物的进入。Arylomycins (8)是一类含有大环的脂肽类抗生素[5], 作用靶标是细菌的Ⅰ型信号肽酶(SPase), SPase是以Ser-Lys为催化二元体(dyad)的丝氨酸蛋白酶, 亲核基团对前手性C=O从S面(S-face)进攻, 生成的手性碳为S构型; 与人体以Ser-His-Asp为催化三元体(triad)的丝氨酸蛋白酶是从R面(R-face)进攻, 生成R构型碳不同, 因而SPase是对人体较小作用的特异性抗菌靶标。阴性菌的SPase处于两层膜之间。Arylomycin具有穿越外膜的性质, 虽然抑制活性较弱(原因是SPaseLepB发生变异降低了结合力), 仍是研制抗阴性菌药物的先导物。
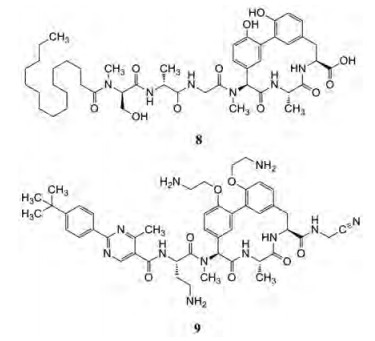
|
分析8与SPaseLepB的共晶结构, 分子中的大环以预制的结构占据了天然配体的结合腔, 所以优化结构不变动大环片段。变化的3个部分是: N-端的脂肪链和三肽片段、两个苯酚基和C端的羧基。经多轮SAR分析, 优化出化合物9 (G0775), 分别用叔丁苯基联嘧啶甲酸和二氨基丁酸代替脂肪链和三肽, 调整了亲脂性并消除了肽的结构; 用氨乙氧基替换容易发生代谢的两个酚羟基, 避免了氧化代谢和葡醛酸苷化, 也提高了溶解性; C端的羧基变成酰胺基乙腈, 分子模拟提示, 氰基接近LepB中参与催化水解的Lysl46, 可以同Lys侧链氨基发生加成反应, 生成脒基的共价化合物如图 13所示, 脒基化合物经LC-MS得到证实。

|
Figure 13 Proposed mechanism of covalent amidine-bond formation between 9 nitrile and lysine 146. The general base (lysine 146) involved in substrate proteolysis functions instead as a nucleophile to attack the nitrile warhead |
化合物9对多株耐药的革兰阴性菌呈现强效活性, MIC为0.125~1.0 μmol·mL-1, 而作为对照化合物8 (arylomycin)的MIC都大于64 μmol·mL-1, 提示9对于SPase发生突变的阴性菌有显著抑制活性。化合物9与突变的LepB共晶结构显示(图 14a和b), 与催化域的20个残基中的8个发生结合作用, 尤其重要的是氰基靠近Lys196, 得以发生不可逆的共价键结合[6]。

|
Figure 14 a. Crystal structure of the protease domain of LepB, with 9 represented as sticks covalently bound to lysine146. b. Overlay of LepB-G0775 and LepB-arylomycin A2 (PDB ID: 1T7D). Comparison of 9 (coloured yellow) and 8 (coloured pink). The catalytic Lys146 is covalently bound to the nitrile warhead, while the serine 91 nucleophile remains unbound. For simplicity, the LepB protein has been removed from each co-structure |
治疗丙型病毒性肝炎的重要靶标是蛋白酶NS3, 属于丝氨酸酶, NS3的功能是裂解结构性蛋白使成熟化, 裂解产物对NS3呈现抑制作用。例如裂解的连接位点NS5A/5BN端生成的产物六肽(10)具有抑制NS3的活性, 该六肽可分为6个域段, 由C端到N端P1~P6, 以此为起始物, 经试探性的修剪和非肽化, 研制丙肝药物。
在结构和计算生物学指引下, 经SAR分析依次消除P6和P5二肽, N端氨基乙酰化, 变换C端为环丙叉甘氨酸和脯氨酸萘甲氧基化得到10a, 继之, 为了提高P3的亲脂性, 将脯氨酸母核变换成环戊二酸, 异丙基变成叔丁基, 萘环修饰成取代的喹啉片段和环丙基的乙烯化成10b, P3片段简化成环戊脲基成10c, 结构生物学提示, 10c环戊基与C端的乙烯基接近, 从而将环戊基与乙烯基融合连接成大环化合物10d, 同时在环丙基处引入磺酰氨基团, 胺基同时连接磺酰基和羰基, 两边拉电子使N-H有弱酸性, 模拟了原来C端的羧基, 并调整了化合物的极性, 用作钠盐; 随后再修饰喹啉环, 得到高活性和选择性的NS3蛋白酶抑制剂11, 即抗丙肝药西米匹韦(simeprevir)。图 15是由六肽演化成西米匹韦的简化流程图, 每一个环节(箭头表示)包含着多轮的合成和构效分析[7-12]。

|
Figure 15 The evolutional scheme of macromolecule simeprevir (11) from hexapeptide (10) |
默沙东研发的伐尼瑞韦(12, vaniprevir)于2018年在日本上市, 是大环化合物抑制NS3蛋白酶的丙肝药物, 经基于结构和分子模拟设计得到。12与上述的西米匹韦有相似的片段, 虽然也是在P2~P4为大环结构, 但结构骨架和大环连接位点不同, 例如12的大环由20个原子组成, 而11是14元环。12的母核为四氢吡咯(羟基脯氨酸), 由1, 3位连接的大环, 芳环参与在大环内, 而11母核是环戊烷, 由1, 2位连出的大环, 芳环没有参与大环的组成[13]。

|
环肽药物大致可分为两类:环肽和钉固肽。环肽是肽链成环状结构, 主要由氨基酸组成的大环化合物; 钉固肽(stapled peptide)是在α螺旋肽链上将隔位的残基用烃基或其他片段相连而形成大环结构的修饰肽。
2.1 环肽药物环肽药物可有不同的结构类型, 完全由氨基酸组成的环肽, 可以是N-端与C-端首尾相连的环肽, 例如环孢菌素(13, cyclosporin A)是由11个氨基酸首尾经肽键相连而形成最大的环, 由于多为N-甲基氨基酸, 对蛋白酶比较稳定, 是少有的可口服环肽。13用作器官移植的排异药物, 然而不良反应较大, 改构的voclosporin (14)只变换一个氨基酸侧链[14]。三期临床表明14的不良反应显著低于环孢菌素A, 而且对狼疮性肾炎有显著疗效。

|
环肽还可以由两个相隔开的半胱氨酸形成二硫键而形成大环, 一些内源性多肽常含有二硫键, 形成稳定构象, 化学性质也稳定。此外, 侧链上的羧基或氨基之间、或与末端的氨基或羧基形成内酰胺, 也可形成大环结构。内源性环肽仍有代谢稳定性问题, 可在不影响大环构象和活性前提下改变个别的氨基酸(非天然, D构型等)提高代谢和/或化学稳定性。
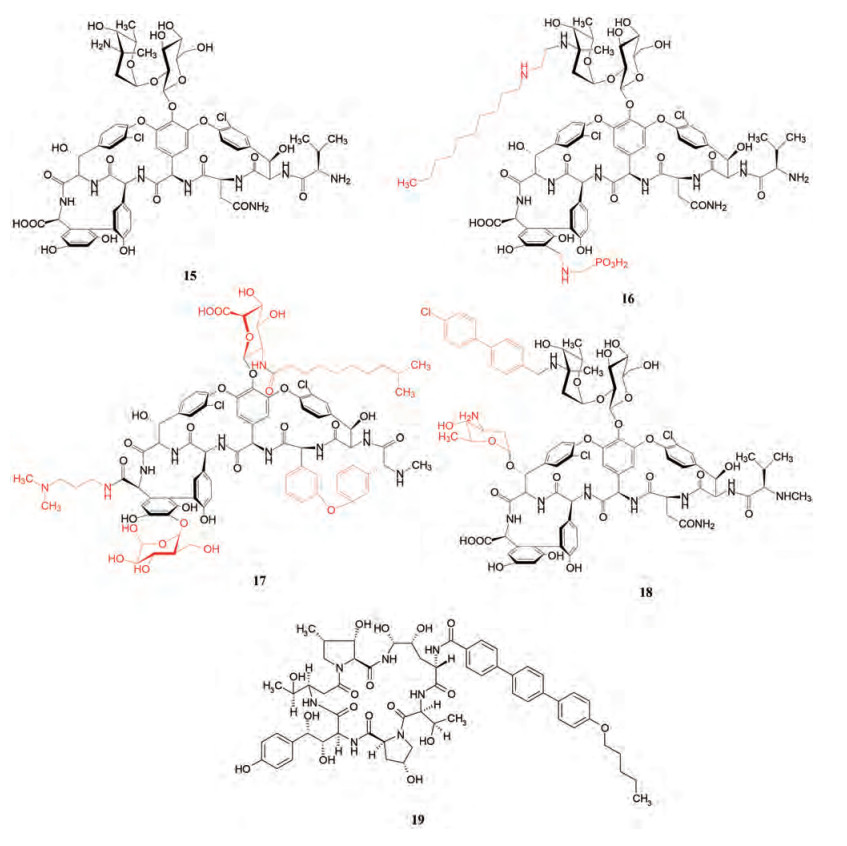
环肽还可与糖片段形成糖肽, 万古霉素(15, vancomycin)是肽链与联苯和二苯醚组成的大环化合物并缀合糖分子而成, 肽片段与细菌合成胞壁前体Lys-D-Ala-D-Ala形成5个氢键结合的网络, 成为强效的抗菌药。为克服万古霉素耐药进行结构修饰, 2009年上市的特拉万星(16, telavancin)和2014年批准的达巴万星(17, dalbavacin)以及奥利万星(18, oritavancin)没有改变环肽的骨架, 只是在环上引入亲脂或亲脂-亲水片段, 增强对耐药菌的敏感性[15]。
抗真菌药物阿尼芬净(19, anidulafungin)是脂环肽药物, 于2006年上市, 为含有正己氧基三联苯的环肽。阿尼芬净是棘球白素(echinocandin)的改构物, 由21元环扩为22元环[16]。
2.2 钉固肽30年前, 研究肽类化合物大都是聚焦于非肽化和肽模拟物(peptidomimetics), 随着对许多蛋白的非可药性(non-druggability)和蛋白-蛋白相互作用(PPI)的深入认识, 以及肽化学和生物药剂学的发展, 直接研制肽类药物已经成为重要领域, 其中钉固肽(stapled peptide)就是近20年来发展的平台技术。
|
PPI是细胞过程和生理机能的重要载体, PPI是在较大范围内发生的结合, 虽然结合有热域(hot spots)之说, 但仍因是在较平坦的界面上作广域结合, 小分子药物干预PPI仍难度很大, 而肽分子可与蛋白有较广泛的结合面, 是研制作用于PPI的活跃领域。
发生PPI最常见的肽为α螺旋, 当肽键的α碳被双取代可提高螺旋的稳定性[17]。若α-螺旋上两个相隔的残基的侧链以共价键形成环桥连接可使螺旋构象稳定化, 这种肽称作钉固肽, 犹如钉固钉对肽作加固。用烃基钉固的α螺旋肽不仅可稳固呈现生物活性的构象, 还能提高对蛋白酶的稳定性, 而且由于是两亲型(amphipathic)分子能够穿透细胞膜, 使得全烃钉固肽不仅能与膜蛋白发生PPI, 而且可进入细胞内发挥作用。此外, 从结构特征分析, 钉固肽与普通肽不同, 它克服了后者未呈结合状态的分子在溶液中多种构象存在, 降低了与靶标蛋白结合重构活性构象所耗费的能量(熵损失), 因而活性提高[18]。α-螺旋肽的每一个螺旋含有3.6个氨基酸残基, 为了在同一侧面上形成钉固装置, 应是在第i、i+4、i+7的位置上形成, 残基侧链上有功能基的氨基酸如Lys、Asp、Cys以及非天然氨基酸之间可按照上述间隔位置形成共价结合的钉固肽。更多的是在拟钉固的两个位置的氨基酸α位预构连接成环的片段, 经特定的化学反应, 偶联成钉固环, 应用最多的是全烃钉固肽[19]。有机合成化学提供了多种催化环合活性的试剂, 图 16是形成不同钉固环的反应类型[20]。

|
Figure 16 Diagram of macrocyclisation chemistries for forming side-chain crosslinks. |
Schafmeister和Verdine最早用合成的方法在预定的位置(例如第i个位置)连接经保护的α-甲基-α-烯烃基甘氨酸, 在第i+n的位置为另一个α-甲基-α-烯烃基甘氨酸(两个烯烃基的大小可以不同), 后经金属络合物催化两个烯基发生偶联反应, 形成i~i+n残基间隔的含有双键的烃基桥环。图 17是合成的示意图。该方法合成的多肽不仅可保持原有的活性, 而且提高了螺旋的结构稳定性和代谢稳定性[21]。

|
Figure 17 Diagram for making all-hydrocarbon stapled peptides |
由于α-甲基-α-烯烃基甘氨酸是手性分子, 铆合在α-螺旋上存在构型问题, 描述这部分的结构表示法, 如图 17中Rii+7S(11)的全烃钉固肽, 表示为从N端到C端的方向, 形成烃基桥的第i个残基为R构型, 第i+7位置的残基结束桥环, 构型为S, 烯烃环含11碳原子。全烃钉固肽犹如在α-螺旋“别卡子”, 加固了α-螺旋的构象而稳定化。大的肽链可以组装两个或多个环烃[22]。
为了提高钉固肽的活性和稳定性, 需要进行分子设计、构效关系和试错试验, 分子设计中有以下诸要素: ①两个钉固残基的距离的优化, 即(i, i+4)或(i, i+7)的选定, 而且肽链上可组装两个或以上的钉固装置; ②两个钉固原子的构型, 可有4种可能: (R, R)、(R, S)、(S, S)和(S, R), 这需要SAR分析确定; ③全烃环的大小(即烃基的大小), 偶联后双键的位置, 双键的E/Z构型等。这几个因素可有不同的组合, 分子模拟可做出预判, SAR加以优化。设计中可用Monte Carlo或分子动力学模拟等计算化学作辅助[20]。
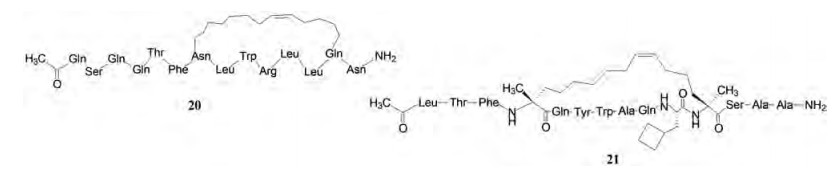
Bernal等[23]研究干预MDM2-p53蛋白-蛋白相互作用, 合成了p5314-29 α-螺旋片段的烃钉固肽, 设计烃基钉固的位置避开与MDM2结合位点, 烃环跨越为i, i+7, 经圆二色谱研究, 合成的8个肽中有的可稳定α-螺旋构象, 其中化合物20与没有烃基钉固的肽比较, 与MDM2结合的α-螺旋构象占85%, 活性Kd = 55 nmol·L-1, 而未钉固化的活性构象只有11%, 活性提高了7倍。此外, 由于钉固化也提高穿越细胞膜的能力。由于保护了天然p53的作用, 癌细胞呈现凋亡作用。
|
第一个进入临床研究的钉固肽是ALRN-6924 (21), 目前在二期阶段, 是p53-MDM2和p53-MDMX的双重抑制剂, 通过噬菌体展示发现的活性多肽, 经第i, i+7的烃基钉固而成的。对MDM2和MDMX结合力Ki分别为0.9和6.8 nmol·L-1。由于阻断了MDM2对p53的结合, 促进了癌细胞的凋亡[24]。
3 结语有机合成反应的进步得以在温和条件下发生C-C键偶联, 形成大环结构, 是研究大环药物的技术支撑。结构生物学、分子模拟和计算化学则指导大环的设计, 从微观结构上设计大环的位置、尺寸和元素组成, 从而开辟了药物创制的新领域, 为研发可药性差的靶标和干预蛋白-蛋白相互作用的药物扩大了化学空间, 钉固肽作为另类大环分子, 虽然仍有许多技术问题尚待解决, 迄今尚无成功的钉固肽上市, 但有潜在的发展优势, 是肽类药物新的生长点, 尤其在作用于细胞因子和信号传导的蛋白-蛋白相互作用中扮演重要的角色。
| [1] |
Giordanetto F, Kihlberg J. Macrocyclic drugs and clinical candidates:what can medicinal chemists learn from their properties?[J]. J Med Chem, 2014, 57: 278-295. DOI:10.1021/jm400887j |
| [2] |
Johnson TW, Richardson PF, Bailey S, et al. Discovery of (10R)-7-amino-12-fluoro-2, 10, 16-trimethyl-15-oxo-10, 15, 16, 17-tetrahydro-2H-8, 4-(metheno)pyrazolo[4, 3-h] [2, 5, 11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1(ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations[J]. J Med Chem, 2014, 57: 4720-4744. DOI:10.1021/jm500261q |
| [3] |
Engelhardt H, Böse D, Petronczki M, et al. Start selective and rigidify:the discovery path toward a next generation of EGFR tyrosine kinase inhibitors[J]. J Med Chem, 2019, 62: 10272-10293. DOI:10.1021/acs.jmedchem.9b01169 |
| [4] |
Corte JR, Fang TA, Osuna H, et al. Structure-based design of macrocyclic factor XIa inhibitors:discovery of the macrocyclic amide linker[J]. J Med Chem, 2017, 60: 1060-1075. DOI:10.1021/acs.jmedchem.6b01460 |
| [5] |
Schimana J, Gebhardt K, Höltzel A, et al. Arylomycins A and B, new biaryl-bridged lipopeptide antibiotics produced by Streptomys sp. Tü 6075. Ⅰ. Taxonomy, fermentation, isolation and biological activities[J]. J Antibiot (Tokyo), 2002, 55: 565-570. DOI:10.7164/antibiotics.55.565 |
| [6] |
Smith PA, Koehler MFT, Girgis HS, et al. Optimized arylomycins are a new class of Gram-negative antibiotics[J]. Nature, 2018, 561: 189-194. DOI:10.1038/s41586-018-0483-6 |
| [7] |
Rancourt J, Cameron D, Gorys V, et al. Peptide-based inhibitors of the hepatitis C virus NS3 protease:structure-activity relationship at the C-terminal position[J]. J Med Chem, 2004, 47: 2511-2522. DOI:10.1021/jm030573x |
| [8] |
Goudreau N, Cameron DR, Bonneau P, et al. NMR structural characterization of peptide inhibitors bound to the HCV NS3 protease:design of a new P2 substituent[J]. J Med Chem, 2004, 47: 123-132. DOI:10.1021/jm0303002 |
| [9] |
Llinàs-Brunet M, Bailey MD, Ghiro E, et al. A systematic approach to the optimization of substrate-based inhibitors of the hepatitis C virus NS3 protease:discovery of potent and specific tripeptide inhibitors[J]. J Med Chem, 2004, 47: 6584-6594. DOI:10.1021/jm0494523 |
| [10] |
Tsantrizos YS, Bolger G, Bonneau P, et al. Macrocyclic inhibitors of the NS3 protease as potential therapeutic agents of hepatitis C virus infection[J]. Angew Chem Int Ed, 2003, 42: 1356-1360. DOI:10.1002/anie.200390347 |
| [11] |
Llinàs-Brunet ML, Bailey MD, Bolger G, et al. Structure-activity study on a novel series of macrocyclic inhibitors of the hepatitis C virus NS3 protease leading to the discovery of BILN 2061[J]. J Med Chem, 2004, 47: 1605-1608. DOI:10.1021/jm0342414 |
| [12] |
Raboisson P, de Kock H, Rosenquist A, et al. Structure-activity relationship study on a novel series of cyclopentane-containing macrocyclic inhibitors of the hepatitis C virus NS3/4A protease leading to the discovery of TMC435350[J]. Bioorg Med Chem Lett, 2008, 18: 4853-4858. DOI:10.1016/j.bmcl.2008.07.088 |
| [13] |
McCauley JA, McIntyre CJ, Rudd MT, et al. Discovery of vaniprevir (MK-7009), a macrocyclic hepatitis C virus NS3/4a protease inhibitor[J]. J Med Chem, 2010, 53: 2443-2463. DOI:10.1021/jm9015526 |
| [14] |
Birsan T, Dambrin C, Freitag DG, et al. The novel calcineurin inhibitor ISA247:a more potent immunosuppressant than cyclosporine in vitro[J]. Transpl Int, 2005, 17: 767-771. |
| [15] |
Bambeke FV. Lipoglycopeptide antibacterial agents in Gram-positive infections:a comparative review[J]. Drugs, 2015, 75: 2073-2095. DOI:10.1007/s40265-015-0505-8 |
| [16] |
Brüggemann RJ, Van Der Velden WJ, Knibbe CA, et al. A rationale for reduced-frequency dosing of anidulafungin for antifungal prophylaxis in immunocompromised patients[J]. J Antimicrob Chemother, 2014, 70: 1166-1174. |
| [17] |
Karle IL, Flippen-Anderson JL, Uma K, et al. Modular design of synthetic protein mimics crystal-structures, assembly, and hydration of two 15- and 16-residue apolar, leucyl-rich helical peptides[J]. J Am Chem Soc, 1990, 112: 9350-9356. DOI:10.1021/ja00181a042 |
| [18] |
Lau YH, De Andrade P, Wu Y, et al. Peptide stapling techniques based on different macrocyclisation chemistries[J]. Chem Soc Rev, 2015, 44: 91-102. DOI:10.1039/C4CS00246F |
| [19] |
Henchey LK, Jochim AL, Arora PS, et al. Contemporary strategies for the stabilization of peptides in the alpha-helical conformation[J]. Curr Opin Chem Biol, 2008, 12: 692-697. DOI:10.1016/j.cbpa.2008.08.019 |
| [20] |
Tan YS, Lane DP, Verma CS. Stapled peptide design:principles and roles of computation[J]. Drug Discov Today, 2016, 21: 1642-1653. DOI:10.1016/j.drudis.2016.06.012 |
| [21] |
Schafmeister CE, Verdine GL. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides[J]. J Am Chem Soc, 2000, 122: 5891-5892. DOI:10.1021/ja000563a |
| [22] |
Walensky LD, Bird GH. Hydrocarbon-stapled peptides:principles, practice, and progress[J]. J Med Chem, 2014, 57: 6275-6288. DOI:10.1021/jm4011675 |
| [23] |
Bernal F, Tyler AF, Korsmeyer SJ, et al. Reactivation of the p53 tumor suppressor pathway a stapled p53 peptide[J]. J Am Chem Soc, 2007, 129: 2456-2457. DOI:10.1021/ja0693587 |
| [24] |
Ng SY, Yoshida N, Christie AL, et al. Targetable vulnerabilities in T- and NK-cell lymphomas identified through preclinical models[J]. Nat Commun, 2018, 9: 2024. DOI:10.1038/s41467-018-04356-9 |