 2020, Vol. 55
2020, Vol. 55

新药创制是复杂的智力活动, 涉及科学研究、技术创造、产品开发和医疗效果等多维科技活动。每个药物都有自身的研发轨迹, 而构建化学结构是最重要的环节, 因为它涵盖了药效、药代、安全性和生物药剂学等多维性质。本栏目以药物化学视角, 对有代表性的药物的成功构建, 加以剖析和解读。
治疗2型糖尿病药物吡格列酮的研发路径与当今以靶标为核心的新药研发不同, 是先确定了疗效, 然后阐明机制和作用靶标, 这是20世纪80年代以前大都沿用的模式, 例如青蒿素的抗疟作用机制至今仍未完全解析, 但不妨碍拯救数百万患者的生命。借助可靠的动物模型和娴熟的药物化学研发技能, 得以使吡格列酮成为新型的抗2型糖尿病的药物。
(编者按)
20世纪70年代日本武田公司着手研究降脂和减肥药, 得益于有遗传性肥胖和糖尿病KK小鼠模型。研发者借鉴已有降脂活性的芳烷酸的化合物结构, 发现了化合物1 (AL-294)具有降低KK小鼠血糖和血脂作用(Kawamatsu Y, Saraie T, Imamiya E, et al. Studies on antihyperlipidemic agents. I. Synthesis and hypolipidemic activities of phenoxyphenyl alkanoic acid derivatives. Arzneimit-Forschung, 1980, 30: 454-459)。虽然那时还不知道作用机制和靶标, 但用整体动物的生理表型变化—血糖和血脂的变化, 评价化合物活性, 与离体靶标相比, 动物模型更接近临床应用的方式。
|
动物为9周龄雄性KK小鼠, 预饲3日粉状饲料, 后用含有1%受试物(重量百分率)的饲料饲养2日(自由饮水), 取静脉血, 测定血中葡萄糖和甘油三酯的浓度。与空白对照比较, 降低70%~89%的活性等级定为4, 50%~69%为3, 30%~49%为2, 10%~29%为1, 低于9%为0。
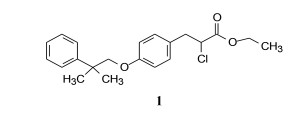
1.3 先导物的初步改造化合物1为取代的苯丙酸酯骨架, 结构中的氯原子为氯苄结构, 也是α-氯代羧酸, 由于与氯原子相连sp3杂化碳具有亲电性, 是容易发生“乱打”靶标的基团, 结构修饰中去除氯原子是必然的。但用氢、羟基、甲氧基或氨基等置换氯原子都使活性降低(Kawamatsu Y, Asakawa H, Saraie T, et al. Studies on antihyperlipidemic agents. Ⅱ. Synthesis and biological activities of 2-chloro-3-arylpropionic acids. Arzneimit-Forrschung, 1980, 30: 585-589), 进而用巯基和硫醚取代, 以及形成杂环(含有氢键给体和接受体)等代替氯乙酸片段, 有代表性的化合物的活性列于表 1。
| Table 1 Activity of the compounds with altering chloropropionic acid moiety |

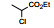

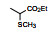

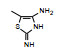
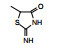

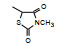

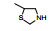


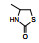
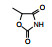
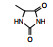
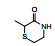
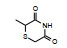
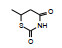
表 1的构效关系提示, 巯基置换氯原子(2)活性减弱, 甲硫基(3)或巯基乙酰化(4)均失去活性。五元杂环噻唑烷类化合物5~15, 活性最强的化合物7 (代码AL-321), 含有噻唑烷二酮环, 由于环上亚胺的氢原子被甲基化后失去活性, 提示该弱酸性氢(两个羰基拉电子效应)可能是结合的重要因素。化合物6和9显示的活性可能是=NH和=S发生水解生成7的缘故。单酮或无酮基化合物基本没有活性。化合物14~18虽然也含有弱酸性氢, 但基本未呈现活性, 说明噻唑烷二酮片段与结合位点呈特异性结合。活泼氢是必要因素, 但不是充分因素(Sonda T, Mizuno K, Tawada H, et al. Studies on antidiabetic agents. Ⅱ. Synthesis of 5-[4-(2-methyl-2-phenylpropoxy)-benzyl]thiazolidine-2, 4-dione (AL-321) and related compounds. Chem Pharm Bull, 1982, 30: 3563-3673)。
2 以噻唑烷二酮为核心的化合物修饰 2.1 外端苯乙基的优化具有噻唑烷二酮药效团的化合物7呈现较强的降糖和降脂活性, 下一步结构优化是固定噻唑烷二酮和p-苄氧结构, 变换外端苯环的取代基(R)和偕甲基(L), 合成的化合物列于表 2。
| Table 2 Activity of compounds with altering terminal phenyl substituents and gem-methyl groups |

表 2的构效关系提示: ①在末端苯环上引入取代基大都降低活性; ②去除苯乙基链上的的偕甲基有利于降糖和降脂活性, 例如化合物27优于7, 而且链上没有偕甲基的结构即使苯环上引入取代基如29、31和35的活性也不错。提示侧链宜简的可能性(Sonda T, Mizuno K, Imamiya E, et al. Studies on antidiabetic agents. Ⅱ. Synthesis of 5-[4-(1-methylcyclohexylmethoxy)-benzyl]thiazolidine-2, 4-dione (ADD-3878) and its derivatives. Chem Pharm Bull, 1982, 30: 3563-3673)。
2.2 侧链长度对活性的影响为了考察苯乙基链长对活性的影响, 合成了不同长度的碳链, 化合物活性列于表 3。
| Table 3 Activity of compounds with varied side chain length |
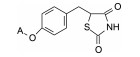
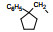
表 3与表 2化合物的两个苯环间链长不同对活性有影响, 以两个碳原子为优选, 例如化合物7、27、41活性强于相隔3或4个碳原子的化合物38和39, 也强于相隔一碳的45或连接苯环的化合物46, 33优于48, 35优于50等。其中化合物27、29、32、35和41降糖和降脂活性都很强, 显著优于7。然而这些化合物在高剂量长期给药时, 动物的肝脏和肝脂肪重量增加, 这种不利的结果可能是结构上存在两个苯环, 诱导代谢所致, 因而探索去除末端苯环, 考察对活性和不良反应的变化。
2.3 脂肪链(环)替换末端苯环苯乙基被脂肪链或脂肪环置换, 表 4列出的化合物表明, 较小取代基置换的化合物呈现良好活性。例如化合物66 (取代的环己基, 环格列酮)有良好的降糖活性, 虽然降脂作用一般, 但安全性优于其他化合物。69 (取代的环戊基)的降糖活性也很高, 但呈现毒性。化合物64的活性和安全性也不错。
| Table 4 Compounds with replacing aliphatic chain/cycle moieties for the phenyl ring |
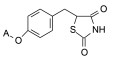
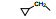
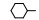
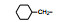
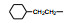

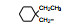


上述的烷氧基化合物具有较强的亲脂性, 为了探索提高极性对活性的影响, 合成了吡啶烷基或胺烷基等化合物。表 5的活性提示73~76有较强的活性, 然而高活性的化合物87、89和90出现一些毒性作用。
| Table 5 Activity of the compounds containing polar groups |
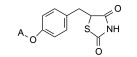


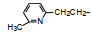




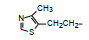

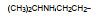
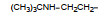
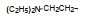


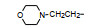
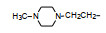

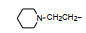
前述与苯环相连的烷基或芳烷基的化合物都是经过氧原子(醚键)连接的, 表 6列出的化合物是考察不经氧原子桥连对活性的影响。结果表明, 没有高活性化合物的出现, 相当多的化合物对降糖和降脂都失去活性, 如化合物95~97、100、102~104和108等, 提示苯环与左侧基团连接的氧原子对与靶标结合的重要性。
| Table 6 Activity of the compounds without attached oxygen atom to the left-hand phenyl ring |





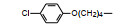


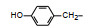
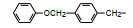
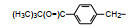
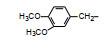

分析上述的近百个化合物的构效关系, 提示p-氧苄基吡咯烷酮是支撑活性化合物的骨架。下一步是将苄基变换为吡啶甲基, 考察环上增添一个氮原子对活性的影响, 表 7的数据表明, 除化合物116和117外活性都减弱, 提示p-氧苄基吡咯烷酮骨架的重要性(Sohda T, Mizuno K, Imamiya E, et al. Studies on antidiabetic agents. Ⅱ. Synthesis of 5-[4-(1-methylcyclohexylmethoxy)-benzyl]thiazolidine-2, 4-dione (ADD-3878) and its derivatives. Chem Pharm Bull, 1982, 30: 3580-3600)。所以氧吡啶甲基的变换不可取。至此, 化合物66由于活性强和不良反应少, 是优良的化合物, 探索进一步开发, 命名为环格列酮(ciglotazone), 进行了深入的研究。
| Table 7 Activity of 2-alkoxypyridyl-5-methylthiazolidinediones |

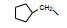

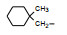


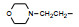

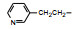

环格列酮进行了临床前研究, 噻唑烷二酮环上有手性碳原子, 拆分成光活体的两个异构体分别评价对KK小鼠的降糖降脂作用, 结果表明二者没有显著差异, 后经犬与猴体内试验证实两个光学异构体在体内迅速发生消旋化。从化学上分析, 手性中心相邻的羰基碳和硫原子的拉电子效应, 可经烯醇化而消旋。

|
环格列酮进入了临床研究, 试验结果没有达到预期的降糖效果, 因而终止了对环格列酮的开发。继续做结构优化。
3 含吡啶环的候选化合物确定 3.1 含吡啶的苯氧乙基骨架的再研究前述的化合物73~77呈现较高的体外活性, 深入测定体内活性, 用不同剂量受试物与饲料混合给糖尿病肥胖KK小鼠, 连续4天后, 测定血糖和血中甘油三酯的含量, 计算降低25%葡萄糖和甘油三酯的剂量, 单位是mg·kg-1·d-1。结果表明74和75显示较高活性, 而73、76和77活性很弱。进一步设计合成了变换吡啶的连接位置(表 8), 提示氮原子在连接的邻位具有活性, 而间位和对位活性很弱。烷基链长仍以2或3个碳原子活性高。
| Table 8 Activity of the compounds with various pyridylalkyloxy moiety. a: Effective dose (mg·kg-1·d-1) of 25% reduction in glucose of diabetic obese KK mice after 4 day's successive oral administration. b: Effective dose (mg·kg-1·d-1) of 25% reduction in triglyceride of diabetic obese KK mice after 4 day's successive oral administration |

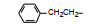


以化合物74为新的起点, 考察吡啶环上的取代基和连接链上取代基对活性的影响, 合成的化合物列于表 9。
| Table 9 Structures and activity of the compounds with various substituents at pyridylalkoxy moiety. a: Effective dose (mg·kg-1·d-1) of 25% reduction in glucose of diabetic obese KK mice after 4 day's successive oral administration. b: Effective dose (mg·kg-1·d-1) of 25% reduction in triglyceride of diabetic obese KK mice after 4 day's successive oral administration |
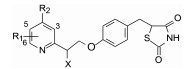
表 9中的构效关系提示, 吡啶的2位侧链的α位引入羟基或羟甲基, 比没有取代的相应化合物活性高, 例如124比74, 126比125, 127比125, 130比129等。然而吡啶环上的羟基或羟甲基取代活性显著降低, 如137和138的活性很低。
对高活性的化合物74、75、126、130~136比较了安全性, 化合物131显示有较高的安全性, 确定作为候选化合物进入开发阶段(Sohda T, Momose Y, Meguro K, et al. Studies on antidiabetic agents. Synthesis and hypoglycemic activity of 5-[4-(pyridylalkoxy)benzyl]-2, 4-thiazolidinediones. Arzneimitt-Forschung, 1990, 45: 37-42)。
3.3 候选化合物的药效学化合物131定名为吡格列酮(pioglitazone), 药效学实验表明, 遗传性肥胖和糖尿病KK小鼠口服2.4~24.5 mg·kg-1·d-1、Zucker肥胖大鼠0.1~10 mg·kg-1·d-1, 连续用药4天, 可显著降低高甘油三酯症、高血脂症、高胰岛素血症和动物对胰岛素抗性的血糖和血脂症。对胰岛素有中等抗性的肥胖型成年比格犬1 mg·kg-1·d-1给药, 可降低空腹下血浆葡萄糖水平, 也减低餐后的脂质水平, 但不影响链佐星(streptozocin)诱导的大鼠的葡萄糖和脂质水平, 提示吡格列酮的作用环节是提高胰岛素对外周(肌肉和脂肪)组织的作用, 对胰岛素抗性的葡萄糖和脂质异常是有效的, 可用于治疗非胰岛素依赖的糖尿病。吡格列酮可口服吸收, 口服生物利用度F=83%, 空腹用药2 h达峰, 半衰期16~24 h, 7天后达稳态(Ikeda H, Taketomi S, Shimura Y, et al. Effects of pioglitazone on glucose and lipid metabolism in normal and insulin resistant animals. Arzneimitt-Forschung, 1990, 57: 156-161), 经临床研究表明是治疗2型糖尿病的有效药物, 于1999年在日本和美国批准上市。
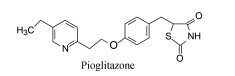
|
以靶标为起点的新药研发, 需要在临床作概念验证(proof of concept), 以动物或细胞表型变化作药物研发, 需要做实验室机制研究(mechanism of action)。研究表明, 吡格列酮是过氧化物酶体增殖因子活化受体γ (PPARγ)激动剂。PPAR属于核受体家族, 有3种亚型: PPARα, β和γ, 分别具有不同的组织分布、结构和功能。PPARγ主要在胰岛β细胞、脂肪组织和血管内皮细胞表达。吡格列酮激活PPARγ, 通过多种机制可改善患者对胰岛素的抵抗, 发挥抗2型糖尿病的作用。
5 安全性吡格列酮体内代谢产物主要是吡啶环上的乙基被氧化成羟基和乙酰基化合物, 以及氧乙基链的氧化脱烷基形成酚基化合物, 而噻唑烷二酮被氧化成亲电性基团的几率较小[这与被撤市停止使用的曲格列酮(139, troglitazone)不同], 加之服用剂量较小(15~20 mg), 因而发生特质性药物毒性(IDT)不明显, 然而长时间大剂量服用吡格列酮有发生膀胱癌的风险。
由于GSK公司研发的另一个PPARγ激动剂罗格列酮(140, rosiglitazone)有引发心血管事件的风险, 已于2008年被要求黑标警示。但2016年报道吡格列酮却有降低心血管事件的效果, 不过在使用说明书上难于表述。

|