 2020, Vol. 55
2020, Vol. 55



ACK1 (活化的Cdc42相关激酶1), 也称为TNK2, 是人类中由TNK2基因编码的酶[1]。ACK1是非受体酪氨酸激酶ACK激酶家族的一员, 最初作为Cdc42蛋白的下游效应蛋白而被鉴定出来, 是Cdc42的靶蛋白。它是目前已知唯一具有SH3结构域的一种非受体酪氨酸激酶, 该结构域位于其激酶结构域羧基末端。
ACK1可与受体酪氨酸激酶(MERK、EGFR等), 癌蛋白AKT[2, 3], 雄激素受体AR[4], 肿瘤抑制因子WWOX[3, 5]、FYN[6]和Grb2[7]等多种蛋白相互作用。它作为癌蛋白在多种肿瘤的发生发展中发挥重要作用。研究结果表明, ACK1与肿瘤细胞的生长增殖密切相关[1]。在前列腺癌、乳腺癌、胰腺癌、肺癌、胃癌[8]、结肠癌[9]、肝癌[3, 10]和卵巢癌等细胞中, 研究者均观察到ACK1的过度激活, 其过度表达与癌症进展和预后有显著相关性。ACK1的激活主要通过其与AKT、AR和KDM3A等下游底物发生相互作用, 进而在底物的特定酪氨酸残基处磷酸化而发挥功能。近来, 以ACK1为靶点的抗肿瘤靶向药物研发多有报道[11-13]。目前, 这类小分子抑制剂多数都处于临床前研究阶段。
1 ACK1的结构ACK1是一种非典型的非受体酪氨酸激酶(NRTK), 包含1 038个氨基酸残基, 大小为143 kDa, 其编码基因TNK2位于3q29染色体区域, 并拥有多个结构域, 其结构域构成在NRTK中独一无二, 是唯一具有CRIB结构域的NRTK, 并且SH3结构域(激酶结构域的C末端)的位置不同于NRTK家族的其他成员。ACK1拥有多个结构域, 参与调节ACK1各种功能, 包括:介导ACK1的膜定位及其二聚化与活化的SAM结构域[14]、酪氨酸激酶活性结构域、通过分子内机制与富含脯氨酸的区域结合发挥自身抑制作用的SH3结构域、介导ACK1与Cdc42的相互作用的CRIB结构域[13]、CBD结构域、介导ACK1与WW结构域相互作用的PPXY基序、介导ACK1与受体酪氨酸激酶相互作用的MHR结构域[15], 以及参与调控ACK1与泛素结合及自身多聚泛素化降解的UBA结构域[16]等。
2 ACK1的激活目前已发现的ACK1激活方式主要有3种, 如图 1所示包括: ①通过多种受体酪氨酸激酶与ACK1相互作用而激活。当细胞受到外界环境中表皮生长因子受体(EGFR)[17]、胰岛素受体、人表皮生长因子受体调节蛋白(heregulin)、血小板衍生的生长因子(PDGFR)、生长特异性蛋白6 (Gas-6)受体、缓激肽, M3毒蕈碱受体和整联蛋白介导的细胞粘附等刺激后, 多种促细胞生长信号通过受体酪氨酸激酶而转导至ACK1[6, 18]。ACK1通常与这些活化的受体结合从而激活自身, 再将相应信号传到下游的效应蛋白。尽管ACK1被多种细胞外信号激活, 但其激活机制及其细胞底物的特性尚未被揭示。② ACK1基因扩增导致其mRNA及蛋白水平升高, 促进其二聚化而激活ACK1。通过基因组测序的相关研究发现, 前列腺癌、肺癌、乳腺癌等多种肿瘤中均有ACK1基因水平的扩增[19], 其过度表达增加了肿瘤的侵袭性[20], 与肿瘤的转移和预后不良相关。③由ACK1基因突变产生异常的激活突变体(表 1)。目前已鉴定出4个与肿瘤相关的ACK1异常活化的体细胞错义突变:在肺腺癌中鉴定出SAM结构域(R34L)的突变; 在卵巢癌中鉴定出激酶催化结构域(E346K)和SH3结构域(R99Q)的突变; 在胃腺癌中发现了SH3结构域(M409I)的突变, 其可防止自身抑制而致ACK1激活失调[1, 2, 19]。另外, 有研究对不同来源的261个癌细胞系和15个正常对照组织的90个酪氨酸激酶基因的转录序列进行测序分析后, 在ACK1基因中发现了10个新的体细胞突变(R127H、A634T、S699N、P731L、R748W、G947D、M393T、H37Y、E111K、S985N)和2个种系突变(P725L、R1038H)。其中的89个肿瘤样本中都出现了P725L突变, 10个样本中都出现了R1038H突变[21, 22]。研究结果显示, 癌症相关突变增加了ACK1自身磷酸化, 而不影响定位和ACK1活性增加, 但增强了ACK1促进肿瘤增殖和迁移的能力, 表明点突变是ACK1失调的机制。

|
Figure 1 ACK1 activation and its role in cancer progress |
| Table 1 Summary of somatic mutations associated with ACK1 aberrant activation in human cells |
总体而言, 几乎四分之一的宫颈癌、卵巢癌和肺癌发生ACK1基因扩增, 而ACK1中的错义替换发生频率明显较低, 这表明基因扩增是大多数癌症中失调的ACK1激活的病因学基础。
3 ACK1的激活在癌症中发挥重要作用关于ACK1在肿瘤中的功能研究已有大量报道。ACK1最早被发现具有促癌作用是发现ACK1的激活可以促进细胞的非贴壁生长与体内肿瘤的生长[23]; 之后的实验表明ACK1是v-Ras转化的细胞生存所必需的[24]。ACK1基因扩增和过表达发生在多种肿瘤类型中, 与不良预后相关, 并与体外细胞运动性和侵袭性增加以及体内转移相关[20, 25]。活化的ACK1表达与肿瘤进展的严重程度呈正相关, 与癌症患者的生存呈负相关[2], 均表明ACK1以多种方式促进肿瘤发生发展。如图 1所示主要有以下几方面:
3.1 ACK1可调节前列腺癌中的雄激素受体(AR)ACK1与AR的相关研究[4]表明, ACK1结合AR并在两个不同的位点Tyr-267和Tyr-363直接磷酸化AR, 两者都位于反式激活结构域内, 促进雄激素非依赖性转录激活AR。ACK1在AR转录起始位点上游的Tyr-88位点磷酸化组蛋白H4, 导致AR转录增加[26]。AR点突变体的表达抑制ACK1驱动的异种移植肿瘤生长。激活的ACK1促进LNCaP和LAPC-4前列腺异种移植肿瘤的雄激素非依赖性生长、雄激素应答增强剂的AR募集以及雄激素缺乏时雄激素诱导的基因表达。Heregulin介导的HER2酪氨酸激酶活化导致ACK1激活和AR磷酸化, ACK1则通过直接磷酸化AR反式激活结构域中的Tyr-267和Tyr-363来促进AR的激活以及AR与雄激素反应元件(ARE)的结合[27]。在AREs处结合的ACK1/AR复合物可导致雄激素诱导的基因表达, 参与调控多种p53依赖性DNA损伤信号通路相关蛋白的转录, 如p300、MDM2和ATM。抑制雄激素不能促进前列腺异种移植肿瘤的雄激素依赖性生长。另外, 最新报道表明, ACK1抑制剂可抑制前列腺癌干细胞的存活[28]。
3.2 ACK1促进乳腺癌迁移与侵袭ACK1在乳腺癌中高表达, ACK1的激活能促进乳腺癌细胞的迁移与侵袭[29]。有研究表明ACK1作为Cdc42的重要下游蛋白, 可以通过SH3结构域与BCAR1 (breast cancer antioestrogen resistance 1)的SH3结构域相互作用, 直接磷酸化其底物结合结构域位点, 从而促进Cdc42依赖性的细胞迁移。即便是在Cdc42 (12V)表达量较低的T47D乳腺癌细胞系中, ACK1的敲低都能显著抑制其迁移能力[30]。在临床上, 活化的ACK1的表达量越高, 相应的乳腺癌患者的预后越差[29]。
3.3 ACK1激活AKT而促进胰腺癌肿瘤生长AKT在胰腺癌中经常被激活, 这与HER-2/neu过度表达相关[31]。ACK1直接与AKT相互作用并使其在176位置的酪氨酸残基磷酸化[1, 2]。Tyr176磷酸化的AKT由于其优先结合磷脂酸(PA)而定位于质膜上, 并在Ser473和Thr308位点进一步磷酸化而激活AKT, 参与抑制细胞凋亡相关基因的表达并促进细胞增殖。Tyr176-磷酸化-AKT和Tyr284-磷酸化-ACK1的表达水平与疾病进展的严重程度呈正相关, 与肿瘤患者的生存率呈负相关[2]。ACK1抑制剂可以使胰腺癌细胞增殖能力明显下降, 并诱导细胞凋亡。表明激活的ACK1可能是确定早期或晚期胰腺癌的预后指标。因此, ACK1抑制剂有望进行治疗性干预以抑制胰腺肿瘤的生长[32]。
3.4 ACK1是慢性白血病中突变型CSF3R的效应因子研究报道采用酪氨酸激酶的特异性siRNA筛选白血病细胞的遗传驱动因子, 发现编码集落刺激因子3 (CSF3R)受体基因突变, 导致下游激酶通过ACK1或JAK激酶的激活, 提示ACK1是慢性中性粒细胞白血病中突变型CSF3R的效应因子, 而这些细胞也表现出对激酶抑制剂的不同敏感性[33]。研究表明, 给予携带JAK活化CSF3R突变的慢性中性白血病(CNL)或非典型CML患者以JAK1/2酪氨酸蛋白激酶抑制剂后, 明显改善临床症状, 但ACK1抑制剂的有效性仍有待观察, ACK1在慢性白血病中的作用尚不明朗。
4 ACK1小分子抑制剂迄今为止, 针对ACK1激酶的高效特异性小分子抑制剂的研发已报道了至少8种(表 2)。本文将分别详细讨论这些小分子抑制剂的有效性及其可能的临床应用前景。
| Table 2 Summary of small molecule inhibitors of ACK1 |
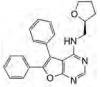

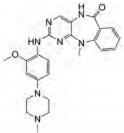
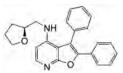
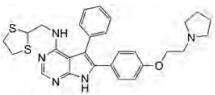
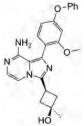
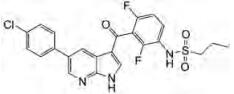
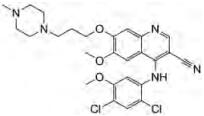
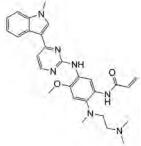
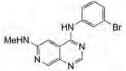
AIM-100是在高通量筛选中发现的第一个也是研究较多的ACK1抑制剂[34, 35]。AIM-100为ATP类似物, 在其浓度不断增加的情况下进行的激酶检测显示, 它可特异性地抑制ACK1, IC50值为21 nmol·L-1, 但不抑制其他30种激酶, 如AKT、AXL、HER、JAK、ERK和PI3K等亚家族的成员[35]。
AIM-100不仅抑制ACK1活化, 还可以在前列腺癌中抑制pTyr267-AR磷酸化以及随后的染色质募集, 抑制AR的促转录活性以及细胞的生长增殖[34], 从而抑制去势和放射抵抗性前列腺异种移植瘤生长[34, 35]。同时AIM-100还可抑制多种癌细胞中AKT的Tyr176位的磷酸化和活化, 导致细胞阻滞在细胞周期G1期, 使癌细胞增殖能力明显下降, 并诱导细胞凋亡。
而在乳腺癌中, Kiran等[36]发现, 他莫昔芬处理乳腺癌细胞后, Heregulin激活的ACK1可以磷酸化ER的辅激活因子上保守的酪氨酸1114位点并调控其活性, 进而上调ER所调控的乳腺肿瘤癌基因HOXA1的转录与表达, 表明ACK1的激活可以通过他莫昔芬介导的对ER的抑制而发挥促癌作用; 并且, 以AIM-100抑制ACK1活性, 则使ER所调控的HOXA1转录受到抑制, 在一定程度上显示了ACK1对他莫昔芬耐药的作用。此外, Buchwald等[37]已经证实ER阳性乳腺癌细胞中ACK1蛋白水平依赖于E2/ER。以他莫昔芬处理乳腺癌细胞后, E2/ER信号传导通路受到抑制, 使靶向ACK1的E3泛素连接酶SIAH表达下降, 导致ACK1泛素化降解受到抑制, 蛋白水平升高而被过度激活, 通过其下游靶蛋白促进乳腺癌发展, 这可能是一个新的他莫昔芬耐药机制。总之, 除ACK1在乳腺癌的发生发展中发挥功能外, ACK1抑制剂在逆转与他莫昔芬耐药方面也具有重要作用。因此, AIM-100有望应用于乳腺癌、前列腺癌、肺癌和胰腺癌的治疗。
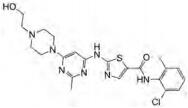
4.2 达沙替尼达沙替尼(Dasatinib)最初被发现为Src和Abl激酶抑制剂[38], 可抑制多种受体和非受体酪氨酸激酶, 因其可抑制Bcr-Abl激酶(包括未突变和突变形式), 故批准用于治疗伊马替尼耐药的慢性粒细胞白血病(CML)[39]。有报道表明, 达沙替尼可以抑制Src FAK介导的信号通路, 从而参与前列腺癌细胞的粘附、迁移、侵袭和转移[40, 41]。达沙替尼还可抑制CaP的生长, 如骨转移[42]、与Src或ACK1对AR的位点特异性酪氨酸磷酸化有关的雄激素非依赖性生长[43], 或CR (CWR22)肿瘤的自发形成[44]。当筛选现有的针对耐药突变激酶的抑制剂时, 发现达沙替尼与ACK1具有高亲和力(Kd = 6 nmol·L-1)[45], 并发现ACK1在肺癌细胞中也可作为潜在的药物靶点[46]。达沙替尼治疗会导致ACK1的4个主要自磷酸化位点(Tyr284、Tyr518、Tyr857和Tyr858)的Tyr-磷酸化显著缺失[46]。
达沙替尼不仅抑制ACK1磷酸化(IC50为1 nmol·L-1), 还可抑制ACK1底物AR的Tyr-267磷酸化。达沙替尼据报道可抑制heregulin诱导的ACK1激酶活性、AR的Tyr-267磷酸化和AR依赖性的报告基因活性, 达沙替尼还可抑制heregulin诱导的内源性AR靶基因的表达。达沙替尼抑制去势小鼠中ACK1依赖性集落的形成和前列腺异种移植肿瘤生长。有趣的是, ACK1或Src敲低时达沙替尼不抑制EGF诱导的AR的Tyr-267磷酸化或EGF激活的AR活性, 提示存在有Tyr-267位点之外的AR磷酸化, 表明特定的酪氨酸激酶激活不同的AR位点磷酸化。达沙替尼可能通过抑制ACK1激酶活性而在前列腺癌中发挥抗肿瘤活性[44]。
4.3 KRCA-0008KRCA-0008又名双-邻-烷氧基-对-哌嗪取代-2, 4-二苯胺基嘧啶, 是一种高效选择性抑制剂, 对ALK和ACK1均具有选择性和有效性, IC50值分别为12和4 nmol·L-1。KRCA-0008在H3122人肺癌小鼠模型中显示了与克唑替尼相当的体内活性[47]。
KRCA-0008具有良好的成药性:良好的水溶性(5% DMSO-水中溶解度为54 μmol·L-1, 5% DMSO-PBS缓冲液中溶解度为150 μmol·L-1); 具有良好的血浆蛋白结合率(大鼠中93%)和在脑中暴露较少(Cbrain/Cplasma = ~0.02);它具有一定的肝微粒体稳定性(30 min后剩余比例分别为:小鼠中52%, 大鼠中89%, 人中72%), 细胞色素P450同工酶(cytochrome P450 proteins, CYP)几乎没有抑制。它几乎不会导致hERG阻滞, 并且在Ames实验、染色体畸变实验和微核实验中均为阴性[48]。KRCA-0008在小鼠和大鼠中也显示出较好的药代动力学性质, 口服生物利用度为66%~94.5%[48], 在人肺癌H3122小鼠模型中具有一定的肿瘤生长抑制作用[49]。KRCA-0008是一种有效的、选择性的和口服生物利用度较高的ACK1抑制剂, 具有较好的临床应用前景。
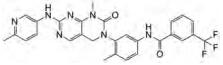
4.4 GNF-7及其衍生物GNF-7又名N-(4-甲基-3-(1-甲基-7-((6-甲基吡啶-3-基)氨基)-2-氧代-1, 4-二氢嘧啶并[4, 5-d]嘧啶-3(2H-基)苯基)-3-(三氟甲基)苯甲酰胺, 是一种多靶点激酶抑制剂, 可作为ACK1和GCK的双重激酶抑制剂, IC50值分别为25和8 nmol·L-1 [50]。GNF-7通过对ACK1/AKT和GCK的联合抑制, 高效并且选择性地抑制了NRAS依赖性的AML和ALL细胞的生长。在移植有人源表达突变型NRAS的MOLT-3-luc+细胞的NSG小鼠中, GNF-7 (15 mg·kg-1, p.o.)显著的下降了疾病的表现, 延长了总体的寿命并且强烈地抑制了P-AKT和P-RPS6的水平[51]。
GNF-7对表达有野生型和突变型Bcr-Abl的Ba/F3细胞中表现了高效的抗增殖活性, 其IC50值均小于11 nmol·L-1。在人源结肠癌细胞系Colo205和SW620中, GNF-7也表现了出色的生长抑制活性, 其IC50分别为5和1 nmol·L-1。同时GNF-7在小鼠中也有较好的药代动力学特性。在移植有T315I-Bcr-Abl-Ba/F3肿瘤的荧光小鼠模型中, GNF-7 (10 mg·kg-1, po)有效地抑制了肿瘤的生长[52]。
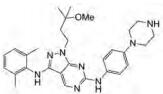
4.5 二芳基吡唑并嘧啶二胺Kopecky等[53]通过分子模拟设计并合成了N3, N6-二芳基-1H-吡唑并[3, 4-d]嘧啶-3, 6-二胺作为非受体酪氨酸激酶ACK1有效和选择性的抑制剂。X射线晶体学研究表明该化合物通过与铰链区的两个氢键和与Thr205的羧基氧的一个氢键靶向到ATP结合部位[53]。尽管该化合物为效果最明显的ACK1抑制剂之一(IC50为20 nmol·L-1), 但它药代动力学特征较差, 降解及血浆清除很快, 而该化合物类似物的口服生物利用度有较显著的改善。该系列化合物目前尚未进行临床实验。
4.6 嘧啶并苯二氮杂䓬Miduturu等[54]针对353种激酶的118种化合物的高通量化合物库筛选, 发现了嘧啶并苯二氮杂䓬类选择性ACK1抑制剂, 其体外Kd为15 nmol·L-1。然而, 需要相对较强剂量的嘧啶并苯二氮杂䓬 (10 μmol·L-1)才能抑制HEK293细胞中EGF诱导的ACK1自身磷酸化。共转染GTP酶缺陷型Cdc42 (Cdc42Q61L突变体), 激活了ACK1激酶, 恢复了部分抑制作用, 表明Cdc42与ACK1的结合可能导致构象变化, 从而降低嘧啶并苯二氮杂䓬的结合[54]。用10 μmol·L-1嘧啶并苯二氮杂䓬处理7天的肺癌细胞数仅减少4倍, 表明该抑制剂抑制癌细胞生长的潜力有限。目前尚处于临床前实验阶段。
4.7 呋喃并嘧啶和吡咯并嘧啶最近的研究通过小分子化合物的高通量筛选揭示了两类新的ACK1抑制剂, 4, 5, 6-三取代的呋喃并[2, 3-d]嘧啶-4-胺(呋喃并嘧啶)和4, 5, 6-三取代的7H-吡咯并[2, 3-d]嘧啶-4-胺(吡咯并嘧啶)[55]。
其中, 呋喃并嘧啶在体外(Ki = 70 nmol·L-1, IC50为5.6 μmol·L-1)能够有效地抑制ACK1活性。但另一抑制剂吡咯并嘧啶二硫杂环戊烷更有效, 在体外(IC50为10 nmol·L-1)表现出更强的抑制活性, 并对ACK1具有更好的选择性(Ki = 0.03 nmol·L-1)。尽管该化合物在细胞水平显示出较强的ACK1抑制作用以及良好的激酶选择性和体外代谢特征, 但药代动力学性质较差, 口服生物利用度低, 且血浆清除速率较快, 降低了其在体内使用的有效性[55]。这两种化合物目前均处于临床前实验阶段。
4.8 咪唑并[1, 5-a]吡嗪衍生物Jin等[56]采用基于药物结构设计和经典药物化学方法, 通过计算机虚拟筛选发现了一系列咪唑并[1, 5-a]吡嗪衍生物可作为ACK1的抑制剂, 根据其衍生物中有效基团的基本结构研究其构效关系、药物代谢和药代动力学(PK)特性。对咪唑并[1, 5-a]吡嗪衍生物进行鉴定, 对代表酪氨酸和丝氨酸、苏氨酸激酶家族的216种纯化蛋白激酶进行分析, 其表现出良好的体内PK特性和选择性[56]。结果表明, 它是一种有效的ACK1抑制剂, 在浓度为1.0 μmol·L-1下, 对ACK1的抑制作用高达100%, 对其他12种激酶(主要包括非受体激酶: YES、FRK、FYN、HCK、LCK、SRC、LYNB、BLK、SRMS、TEC、TXK和FGR等)的抑制作用大于80%。理化性质和ADMET研究也表明咪唑并[1, 5-a]吡嗪衍生物具有良好的类药性质, 提示它是一种有效的选择性ACK1抑制剂, 口服生物利用度高[56], 具有较好的临床应用前景。
4.9 具ACK1抑制活性的其他药物 4.9.1 Vemurafenib (PLX-4032, RG7204或RO5185426)Vemurafenib, 也称为PLX4032、RG7204或RO5185426, 是口服生物可用的, 具有ATP竞争性的BRAF (V600E)激酶小分子抑制剂, IC50为31 nmol·L-1, 具有潜在的抗肿瘤活性。Vemurafenib于2011年8月17日获得FDA批准用于晚期黑色素瘤的治疗[57]。Vemurafenib选择性结合BRAF (V600E)激酶的ATP结合位点并抑制其活性, 从而导致该激酶表达的肿瘤细胞中过度激活的MAPK下游信号通路的抑制, 进而减少肿瘤细胞的增殖[58, 59]。Vemurafenib也抑制一些非-RAF激酶, 包括ACK1、KHS1和SRMS, 其中对于ACK1的IC50为19 nmol·L-1 [60]。由于Vemurafenib是FDA批准的小分子抑制剂, 对于ACK1高表达的肿瘤可能是一种较好的潜在治疗策略。然而, 其在体内抑制ACK1激酶活性的功能尚未进行评估。
4.9.2 Bosutinib Bosutinib (SKI-606)是由辉瑞公司开发的第三代的双SRC-ABL激酶抑制剂, 已被美国食品和药物管理局(FDA)批准用于治疗CML患者[61, 62]。IC50分别为1.2和1 nmol·L-1。Bosutinib作用于Src比非Src家族激酶选择性高, IC50为1.2 nmol·L-1, 且有效抑制Src依赖的细胞增殖, IC50为100 nmol·L-1 [63]。除此之外, 它在KRAS突变的非小细胞肺癌中通过ACK1抑制迁移和侵袭, 能在2.7 nmol·L-1的IC50下结合并阻止ACK1的自磷酸化。在肺癌细胞系和斑马鱼转移模型中观察到, Bosutinib是通过ACK1以KRAS依赖的方式来抑制癌细胞的迁移和侵袭[64]。
4.9.3 AZD9291 (Osimertinib)AZD9291是一种口服给药的不可逆的、突变选择性的第三代表皮生长因子受体酪氨酸激酶(EGFR-TKI)抑制剂, 可选择性抑制EGFR-TKI致敏和EGFR T790M耐药性突变[65, 66], 对L858R和L858R/T790M突变的EGFR活性抑制的IC50分别为12和1 nmol·L-1, 主要用于治疗晚期非小细胞肺癌(NSCLC)[67]。AZD9291对EGFR T790M突变型非小细胞肺癌具有高度选择性[68]。AZD9291在体外和体内均可诱导自噬, 降低EGFR水平, 并通过PI3K/Akt信号通路对NSCLC表现出强烈的抑制作用[69]。此外, AZD9291对其他多种激酶有较好的抑制效果, 如ErbB2/4、ACK1、ALK、BLK、BRK、MLK1、MNK2, 其中对ACK1的IC50为71 nmol·L-1。且AZD9291具有良好的生物利用度, 广泛分布于组织中, 血浆清除率尚可, 小鼠口服半衰期约为3 h[70]。
4.9.4 PD158780PD158780是一种针对EGFR家族均具有活性的抑制剂, 对EGFR、ErbB2、ErbB3和ErbB4的IC50值分别为8 μmol·L-1、49 nmol·L-1、52 nmol·L-1、52 nmol·L-1。PD158780在A431人表皮样癌中可抑制EGFR的自身磷酸化(IC50 = 13 nmol·L-1), 还可以抑制SK-BR-3和MDAMB-453乳腺癌中heregulin的磷酸化, IC50值分别为49和52 nmol·L-1, 表明该化合物对EGFR受体家族的其他成员也具有活性[71]。此外, PD158780也可以抑制ACK1, 在体外IC50约为200 nmol·L-1, 且PD158780对ACK1的抑制作用呈剂量依赖性。但在体内抑制ACK1激酶活性的功能尚未进行评估, 仍需更多深入的临床前研究[24]。
以上几种药物均具有一定的抑制ACK1活性, 但尚缺乏对该抑制作用的深入研究, 以及体内疗效和临床评估, 或许未来也可作为ACK1的选择性抑制剂加以阐释与研发。
5 展望迄今已发现了超过20种与ACK1相互作用的蛋白, 它作为癌蛋白, 主要通过下游底物AKT[1, 2]、AR[4]和KDM3A[36]等过度激活, 在多种肿瘤的发生发展中发挥重要作用。但ACK1是否存在其他机制促进癌细胞生长增殖与迁移侵袭还并不清楚, 更多ACK1相互作用蛋白或底物也亟待鉴定。
前期研究表明[13], ACK1抑制剂在多种肿瘤中可抑制肿瘤生长转移, 具有广阔的临床应用前景, 同时ACK1的T284-磷酸化作为标志物也具有重要的临床诊断价值。由于目前开发的ACK1抑制剂大多存在血浆清除速率较快、口服生物利用度较差等问题[53, 55], 因此, 这也为研发临床上有效的ACK1抑制剂带来更大的机遇和挑战。
迄今为止, 文献中报道的ACK1抑制剂都是ATP类似物, 大多数小分子激酶抑制剂都以激酶的ATP结合位点为靶点[72], 然而ATP竞争性抑制剂(I型抑制剂)通常不能有效区分多种激酶的ATP结合位点[73]。这可能会限制其临床应用, 并增加其因脱靶引起毒性的可能性。由于ACK1是一类多结构域的大分子蛋白, 域间相互作用对其激酶的激活至关重要, 因此, 当小分子或多肽与位于细胞内的变构位点结合时, 可以通过改变活性位点的形状来实现对ACK1激酶结构域的非竞争性抑制, 甚至影响邻近的Sam、Sh3或Crib结构域, 从而达到对ACK1的功能抑制。作为一种个体化治疗策略, ACK1抑制剂的特异性不仅对细胞生物化学的研究非常重要, 而且对抑制肿瘤生长也非常重要。因此, 以激酶的特异性调控位点为靶点, 寻找非竞争性或纯变构抑制剂(Ⅱ-Ⅲ型抑制剂), 可为设计研发ACK1的选择性抑制剂提供更好的机会[74]。尽管, 目前为止还没有获得ACK1特异性变构抑制剂, 利用亲和力选择质谱(AS-MS)方法已成为鉴定其潜在变构抑制剂的良好筛选技术[75], 开发获得新型有效的ATP竞争性和变构ACK1抑制剂, 并最小化其脱靶效应, 减少毒性。故而, 研发ACK1变构抑制剂, 有望成为一种新型的ACK1抑制剂。
相信随着科学技术的进步及其作用机制的深入研究, 研发更多的新型高效ACK1抑制剂, 进一步突破目前面临的瓶颈, 将使其成为治疗肿瘤的有效手段, 并与其他药物联合治疗将可能发挥更好的疗效。例如, 未来利用AIM-100或达沙替尼等抑制剂与其他药物的联合用药, 可能逆转他莫昔芬治疗乳腺癌耐药的发生[36, 37]。当然, 研发针对ACK1的其他类型的靶向药物如抗体制剂、基因治疗等, 也同样需要关注, 但在本综述中不再赘述。
总之, 针对ACK1研发更多更有效的ACK1小分子抑制剂, 并合理应用于临床, 将为肿瘤研究和临床治疗提供一定的科学依据, 具有十分重要的意义。
| [1] |
Mahajan K, Mahajan NP. Shepherding AKT and androgen receptor by ACK1 tyrosine kinase[J]. J Cell Physiol, 2010, 224: 327-333. DOI:10.1002/jcp.22162 |
| [2] |
Mahajan K, Coppola D, Challa S, et al. Ack1 mediated AKT/PKB tyrosine 176 phosphorylation regulates its activation[J]. PLoS One, 2010, 5: e9646. DOI:10.1371/journal.pone.0009646 |
| [3] |
Xie B, Zen Q, Wang X, et al. ACK1 promotes hepatocellular carcinoma progression via downregulating WWOX and activating AKT signaling[J]. Int J Oncol, 2015, 46: 2057-2066. DOI:10.3892/ijo.2015.2910 |
| [4] |
Mahajan NP, Liu Y, Majumder S, et al. Activated Cdc42-associated kinase ACK1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation[J]. Proc Natl Acad Sci U S A, 2007, 104: 8438-8443. DOI:10.1073/pnas.0700420104 |
| [5] |
Mahajan NP, Whang YE, Mohler JL, et al. Activated tyrosine kinase ACK1 promotes prostate tumorigenesis:role of Ack1 in polyubiquitination of tumor suppressor Wwox[J]. Cancer Res, 2005, 65: 10514-10523. DOI:10.1158/0008-5472.CAN-05-1127 |
| [6] |
Linseman DA, Heidenreich KA, Fisher SK. Stimulation of M3 muscarinic receptors induces phosphorylation of the Cdc42 effector activated Cdc42Hs-associated kinase-1via a Fyn tyrosine kinase signaling pathway[J]. J Biol Chem, 2001, 276: 5622-5628. DOI:10.1074/jbc.M006812200 |
| [7] |
Kato-Stankiewicz J, Ueda S, Kataoka T, et al. Epidermal growth factor stimulation of the ACK1/Dbl pathway in a Cdc42 and Grb2-dependent manner[J]. Biochem Biophys Res Commun, 2001, 284: 470-477. DOI:10.1006/bbrc.2001.5004 |
| [8] |
Xu SH, Huang JZ, Chen M, et al. Amplification of ACK1 promotes gastric tumorigenesis via ECD-dependent p53 ubiquitination degradation[J]. Oncotarget, 2017, 8: 12705-12716. DOI:10.18632/oncotarget.6194 |
| [9] |
Lv C, Zhao X, Gu H, et al. Involvement of activated Cdc42 kinase1 in colitis and colorectal neoplasms[J]. Med Sci Monit, 2016, 22: 4794-4802. DOI:10.12659/MSM.902274 |
| [10] |
Lei X, Li YF, Chen GD, et al. ACK1 overexpression promotes metastasis and indicates poor prognosis of hepatocellular carcinoma[J]. Oncotarget, 2015, 6: 40622-40641. DOI:10.18632/oncotarget.5872 |
| [11] |
Lawrence HR, Mahajan K, Luo Y, et al. Development of novel ACK1/TNK2 inhibitors using a fragment-based approach[J]. J Med Chem, 2015, 58: 2746-2763. DOI:10.1021/jm501929n |
| [12] |
Mahajan K, Mahajan NP. PI3K-independent AKT activation in cancers:a treasure trove for novel therapeutics[J]. J Cell Physiol, 2012, 227: 3178-3184. DOI:10.1002/jcp.24065 |
| [13] |
Mahajan K, Mahajan NP. ACK1 tyrosine kinase:targeted inhibition to block cancer cell proliferation[J]. Cancer Lett, 2013, 338: 185-192. DOI:10.1016/j.canlet.2013.04.004 |
| [14] |
Prieto-Echagüe V, Gucwa A, Brown DA, et al. Regulation of ACK1 localization and activity by the amino-terminal SAM domain[J]. BMC Biochem, 2010, 11: 42. DOI:10.1186/1471-2091-11-42 |
| [15] |
Pao-Chun L, Chan PM, Chan W, et al. Cytoplasmic ACK1 interaction with multiple receptor tyrosine kinases is mediated by Grb2:an analysis of ACK1 effects on Axl signaling[J]. J Biol Chem, 2009, 284: 34954-34963. DOI:10.1074/jbc.M109.072660 |
| [16] |
Mahajan K, Mahajan NP. ACK1/TNK2 tyrosine kinase:molecular signaling and evolving role in cancers[J]. Oncogene, 2015, 34: 4162-4167. DOI:10.1038/onc.2014.350 |
| [17] |
Lin Q, Wang J, Childress C, et al. The activation mechanism of ACK1(activated Cdc42-associated tyrosine kinase 1)[J]. Biochem J, 2012, 445: 255-264. DOI:10.1042/BJ20111575 |
| [18] |
Galisteo ML, Yan Y, Ure & ntilde; a J, et al. Activation of the nonreceptor protein tyrosine kinase Ack by multiple extracellular stimuli[J]. Proc Natl Acad Sci U S A, 2006, 103: 9796-9801. DOI:10.1073/pnas.0603714103 |
| [19] |
Prieto-Echagüe V, Gucwa A, Craddock BP, et al. Cancer-associated mutations activate the nonreceptor tyrosine kinase Ack1[J]. J Biol Chem, 2010, 285: 10605-10615. DOI:10.1074/jbc.M109.060459 |
| [20] |
van der Horst EH, Degenhardt YY, Strelow A, et al. Metastatic properties and genomic amplification of the tyrosine kinase gene ACK1[J]. Proc Natl Acad Sci U S A, 2005, 102: 15901-15906. DOI:10.1073/pnas.0508014102 |
| [21] |
Chua BT, Lim SJ, Tham SC, et al. Somatic mutation in the ACK1 ubiquitin association domain enhances oncogenic signaling through EGFR regulation in renal cancer derived cells[J]. Mol Oncol, 2010, 4: 323-334. DOI:10.1016/j.molonc.2010.03.001 |
| [22] |
Ruhe JE, Streit S, Hart S, et al. Genetic alterations in the tyrosine kinase transcriptome of human cancer cell lines[J]. Cancer Res, 2007, 67: 11368-11376. DOI:10.1158/0008-5472.CAN-07-2703 |
| [23] |
Feldman BJ, Feldman D. The development of androgen-independent prostate cancer[J]. Nat Rev Cancer, 2001, 1: 34-45. DOI:10.1038/35094009 |
| [24] |
Nur-E-Kamal A, Zhang A, Keenan SM, et al. Requirement of activated Cdc42-associated kinase for survival of v-Ras-transformed mammalian cells[J]. Mol Cancer Res, 2005, 3: 297-305. DOI:10.1158/1541-7786.MCR-04-0152 |
| [25] |
Modzelewska K, Newman LP, Desai R, et al. ACK1 mediates Cdc42-dependent cell migration and signaling to p130Cas[J]. J Biol Chem, 2006, 281: 37527-37535. DOI:10.1074/jbc.M604342200 |
| [26] |
Mahajan K, Malla P, Lawrence HR, et al. ACK1/TNK2 regulates histone H4 Tyr88-phosphorylation and AR gene expression in castration-resistant prostate cancer[J]. Cancer Cell, 2017, 31: 790-803. DOI:10.1016/j.ccell.2017.05.003 |
| [27] |
Karaca M, Liu Y, Zhang Z, et al. Mutation of androgen receptor N-terminal phosphorylation site Tyr-267 leads to inhibition of nuclear translocation and DNA binding[J]. PLoS One, 2015, 10: e0126270. DOI:10.1371/journal.pone.0126270 |
| [28] |
Mahajan NP, Coppola D, Kim J, et al. Blockade of ACK1/TNK2 To squelch the survival of prostate cancer stem-like cells[J]. Sci Rep, 2018, 8: 1954. DOI:10.1038/s41598-018-20172-z |
| [29] |
Wu X, Zahari MS, Renuse S, et al. The non-receptor tyrosine kinase TNK2/ACK1 is a novel therapeutic target in triple negative breast cancer[J]. Oncotarget, 2017, 8: 2971-2983. DOI:10.18632/oncotarget.13579 |
| [30] |
Howlin J, Rosenkvist J, Andersson T. TNK2 preserves epidermal growth factor receptor expression on the cell surface and enhances migration and invasion of human breast cancer cells[J]. Breast Cancer Res, 2008, 10: R36. DOI:10.1186/bcr2087 |
| [31] |
Manning BD, Cantley LC. AKT/PKB signaling:navigating downstream[J]. Cell, 2007, 129: 1261-1274. DOI:10.1016/j.cell.2007.06.009 |
| [32] |
Mahajan K, Coppola D, Chen YA, et al. ACK1 tyrosine kinase activation correlates with pancreatic cancer progression[J]. Am J Pathol, 2012, 180: 1386-1393. DOI:10.1016/j.ajpath.2011.12.028 |
| [33] |
Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML[J]. N Engl J Med, 2013, 368: 1781-1790. DOI:10.1056/NEJMoa1214514 |
| [34] |
Mahajan K, Challa S, Coppola D, et al. Effect of ACK1 tyrosine kinase inhibitor on ligand-independent androgen receptor activity[J]. Prostate, 2010, 70: 1274-1285. |
| [35] |
Mahajan K, Coppola D, Rawal B, et al. ACK1-mediated androgen receptor phosphorylation modulates radiation resistance in castration-resistant prostate cancer[J]. J Biol Chem, 2012, 287: 22112-22122. DOI:10.1074/jbc.M112.357384 |
| [36] |
Mahajan K, Lawrence HR, Lawrence NJ, et al. ACK1 tyrosine kinase interacts with histone demethylase KDM3A to regulate the mammary tumor oncogene HOXA1[J]. J Biol Chem, 2014, 289: 28179-28191. DOI:10.1074/jbc.M114.584425 |
| [37] |
Buchwald M, Pietschmann K, Brand P, et al. SIAH ubiquitin ligases target the nonreceptor tyrosine kinase ACK1 for ubiquitinylation and proteasomal degradation[J]. Oncogene, 2013, 32: 4913-4920. DOI:10.1038/onc.2012.515 |
| [38] |
Scott AJ, Song EK, Bagby S, et al. Evaluation of the efficacy of dasatinib, a Src/Abl inhibitor, in colorectal cancer cell lines and explant mouse model[J]. PLoS One, 2017, 12: e0187173. DOI:10.1371/journal.pone.0187173 |
| [39] |
Brave M, Goodman V, Kaminskas E, et al. Sprycel for chronic myeloid leukemia and philadelphia chromosome-positive acute lymphoblastic leukemia resistant to or intolerant of imatinib mesylate[J]. Clin Cancer Res, 2008, 14: 352-359. DOI:10.1158/1078-0432.CCR-07-4175 |
| [40] |
Nam S, Kim D, Cheng JQ, et al. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells[J]. Cancer Res, 2005, 65: 9185-9189. DOI:10.1158/0008-5472.CAN-05-1731 |
| [41] |
Rice L, Lepler S, Pampo C, et al. Impact of the SRC inhibitor dasatinib on the metastatic phenotype of human prostate cancer cells[J]. Clin Exp Metastasis, 2012, 29: 133-142. DOI:10.1007/s10585-011-9436-2 |
| [42] |
Koreckij T, Nguyen H, Brown LG, et al. Dasatinib inhibits the growth of prostate cancer in bone and provides additional protection from osteolysis[J]. Br J Cancer, 2009, 101: 263-268. DOI:10.1038/sj.bjc.6605178 |
| [43] |
Liu Y, Karaca M, Zhang Z, et al. Dasatinib inhibits site-specific tyrosine phosphorylation of androgen receptor by Ack1 and Src kinases[J]. Oncogene, 2010, 29: 3208-3216. DOI:10.1038/onc.2010.103 |
| [44] |
Su B, Gillard B, Gao L, et al. Src controls castration recurrence of CWR22 prostate cancer xenografts[J]. Cancer Med, 2013, 2: 784-792. DOI:10.1002/cam4.144 |
| [45] |
Carter TA, Wodicka LM, Shah NP, et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases[J]. Proc Natl Acad Sci U S A, 2005, 102: 11011-11016. DOI:10.1073/pnas.0504952102 |
| [46] |
Li J, Rix U, Fang B, et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer[J]. Nat Chem Biol, 2010, 6: 291-299. DOI:10.1038/nchembio.332 |
| [47] |
Park CH, Choe H, Jang IY, et al. Novel bis-ortho-alkoxy-para-piperazinesubstituted-2, 4-dianilinopyrimidines (KRCA-0008) as potent and selective ALK inhibitors for anticancer treatment[J]. Bioorg Med Chem Lett, 2013, 23: 6192-6196. DOI:10.1016/j.bmcl.2013.08.090 |
| [48] |
Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2, 4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials[J]. J Med Chem, 2013, 56: 5675-5690. DOI:10.1021/jm400402q |
| [49] |
Kang CH, Yun JI, Lee K, et al. Development of potent ALK inhibitor and its molecular inhibitory mechanism against NSCLC harboring EML4-ALK proteins[J]. Biochem Biophys Res Commun, 2015, 464: 762-767. DOI:10.1016/j.bbrc.2015.07.027 |
| [50] |
Cho H, Shin I, Ju E, et al. First SAR study for overriding NRAs mutant driven acute myeloid leukemia[J]. J Med Chem, 2018, 61: 8353-8373. DOI:10.1021/acs.jmedchem.8b00882 |
| [51] |
Nonami A, Sattler M, Weisberg E, et al. Identification of novel therapeutic targets in acute leukemias with NRAS mutations using a pharmacologic approach[J]. Blood, 2015, 125: 3133-3143. DOI:10.1182/blood-2014-12-615906 |
| [52] |
Choi HG, Ren P, Adrian F, et al. A type-II kinase inhibitor capable of inhibiting the T315I "gatekeeper" mutant of Bcr-Abl[J]. J Med Chem, 2010, 53: 5439-5448. DOI:10.1021/jm901808w |
| [53] |
Kopecky DJ, Hao X, Chen Y, et al. Identification and optimization of N3, N6-diaryl-1H-pyrazolo[J]. Bioorg Med Chem Lett, 2008, 18: 6352-6356. DOI:10.1016/j.bmcl.2008.10.092 |
| [54] |
Miduturu CV, Deng X, Kwiatkowski N, et al. High-throughput kinase profiling:a more efficient approach toward the discovery of new kinase inhibitors[J]. Chem Biol, 2011, 18: 868-879. DOI:10.1016/j.chembiol.2011.05.010 |
| [55] |
Jiao X, Kopecky DJ, Liu J, et al. Synthesis and optimization of substituted furo[J]. Bioorg Med Chem Lett, 2012, 22: 6212-6217. DOI:10.1016/j.bmcl.2012.08.020 |
| [56] |
Jin M, Wang J, Kleinberg A, et al. Discovery of potent, selective and orally bioavailable imidazo[J]. Bioorg Med Chem Lett, 2013, 23: 979-984. DOI:10.1016/j.bmcl.2012.12.042 |
| [57] |
Liszkay G. Vemurafenib (Zelboraf) in the therapy of melanoma[J]. Magy Onkol, 2013, 57: 110-113. |
| [58] |
Garbe C, Eigentler TK. Vemurafenib[J]. Recent Results Cancer Res, 2018, 211: 77-89. |
| [59] |
Martin-Liberal J, Larkin J. Vemurafenib for the treatment of BRAF mutant metastatic melanoma[J]. Future Oncol, 2015, 11: 579-589. DOI:10.2217/fon.14.252 |
| [60] |
Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma[J]. Nature, 2010, 467: 596-599. DOI:10.1038/nature09454 |
| [61] |
Stansfield L, Hughes TE, Walsh-Chocolaad TL. Bosutinib:a second-generation tyrosine kinase inhibitor for chronic myelogenous leukemia[J]. Ann Pharmacother, 2013, 47: 1703-1711. DOI:10.1177/1060028013503124 |
| [62] |
Breccia M, Binotto G. Bosutinib for chronic myeloid leukemia[J]. Rare Cancers Ther, 2015, 3: 35-46. DOI:10.1007/s40487-015-0010-y |
| [63] |
Bieerkehazhi S, Chen Z, Zhao Y, et al. Novel Src/Abl tyrosine kinase inhibitor bosutinib suppresses neuroblastoma growth via inhibiting Src/Abl signaling[J]. Oncotarget, 2017, 8: 1469-1480. DOI:10.18632/oncotarget.13643 |
| [64] |
Tan DS, Haaland B, Gan JM, et al. Bosutinib inhibits migration and invasion via ACK1 in KRAS mutant non-small cell lung cancer[J]. Mol Cancer, 2014, 13: 13. DOI:10.1186/1476-4598-13-13 |
| [65] |
Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer[J]. N Engl J Med, 2018, 378: 113-125. DOI:10.1056/NEJMoa1713137 |
| [66] |
Yang JC, Ahn MJ, Kim DW, et al. Osimertinib in pretreated T790M-positive advanced non-small-cell lung cancer:AURA study phase Ⅱ extension component[J]. J Clin Oncol, 2017, 35: 1288-1296. DOI:10.1200/JCO.2016.70.3223 |
| [67] |
Jiang T, Zhou C. Clinical activity of the mutant-selective EGFR inhibitor AZD9291 in patients with EGFR inhibitor-resistant non-small cell lung cancer[J]. Transl Lung Cancer Res, 2014, 3: 370-372. |
| [68] |
Huang L, Huang H, Zhou XP, et al. Osimertinib or EGFR-TKIs/chemotherapy in patients with EGFR-mutated advanced nonsmall cell lung cancer:a meta-analysis[J]. Medicine (Baltimore), 2019, 98: e17705. DOI:10.1097/MD.0000000000017705 |
| [69] |
Zhang Z, Zhang M, Liu H, et al. AZD9291 promotes autophagy and inhibits PI3K/Akt pathway in NSCLC cancer cells[J]. J Cell Biochem, 2019, 120: 756-767. DOI:10.1002/jcb.27434 |
| [70] |
Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer[J]. Cancer Discov, 2014, 4: 1046-1061. DOI:10.1158/2159-8290.CD-14-0337 |
| [71] |
Fry DW, Nelson JM, Slintak V, et al. Biochemical and antiproliferative properties of 4-[J]. Biochem Pharmacol, 1997, 54: 877-887. DOI:10.1016/S0006-2952(97)00242-6 |
| [72] |
Bogoyevitch MA, Fairlie DP. A new paradigm for protein kinase inhibition:blocking phosphorylation without directly targeting ATP binding[J]. Drug Discov Today, 2007, 12: 622-633. DOI:10.1016/j.drudis.2007.06.008 |
| [73] |
Fischer PM. The design of drug candidate molecules as selective inhibitors of therapeutically relevant protein kinases[J]. Curr Med Chem, 2004, 11: 1563-1583. DOI:10.2174/0929867043365062 |
| [74] |
Cox KJ, Shomin CD, Ghosh I. Tinkering outside the kinase ATP box:allosteric (type Ⅳ) and bivalent (type Ⅴ) inhibitors of protein kinases[J]. Future Med Chem, 2011, 3: 29-43. DOI:10.4155/fmc.10.272 |
| [75] |
Comess KM, Trumbull JD, Park C, et al. Kinase drug discovery by affinity selection/mass spectrometry (ASMS):application to DNA damage checkpoint kinase Chk1[J]. J Biomol Screen, 2006, 11: 755-764. DOI:10.1177/1087057106289972 |