 2020, Vol. 55
2020, Vol. 55



西尼罗病毒(West Nile virus, WNV)是一种近年来在全球广泛传播的黄病毒, 为单股线型正链RNA病毒, 人感染后可导致西尼罗热。因1937年首次在乌干达西尼罗地区的一位女性发热患者体内发现而被命名为西尼罗病毒, 此后在亚洲、欧洲和澳大利亚小规模传播, 于1999年在美国爆发并迅速传播到北美其他地区后引起高度关注[1]。迄今为止, 并没有特异性治疗西尼罗病毒感染的上市药物。因此, 研发低毒、高效的抗西尼罗病毒药物具有重要意义。本文精选了近年来西尼罗病毒抑制剂的研发实例, 从药物化学视角总结了西尼罗病毒抑制剂研究的新进展。
1 西尼罗病毒的结构、复制周期与临床表现 1.1 西尼罗病毒结构在低温电子显微镜下观察到病毒颗粒呈球形, 直径在40~60 nm之间。最外层为病毒包膜, 上面镶嵌着膜蛋白, 包膜内是核衣壳, 中心是病毒RNA (图 1)。西尼罗病毒基因组(图 2)是单股线型的正链RNA (即mRNA), 长度约为11 kb, 包括5′端未翻译区、一个长的开放阅读框和3′端未翻译区。开放阅读框编码一个多聚蛋白, 该多聚蛋白在病毒蛋白酶(NS2B/NS3蛋白酶)及宿主蛋白的共同作用下, 被切割成3个结构蛋白(C、prM、E)和7个非结构蛋白(NS1、NS2A、NS2B、NS3、NS4A、NS4B和NS5)。结构蛋白主要参与病毒颗粒的形成过程, 而非结构蛋白主要负责病毒复制、病毒粒子组装和逃避宿主先天免疫反应。E蛋白介导病毒和宿主细胞膜融合使病毒进入宿主细胞。成熟的病毒颗粒中含有M蛋白, 它是由弗林(Furin)蛋白酶裂解prM蛋白产生的, 这是病毒成熟的关键环节。病毒粒子内部是C蛋白和RNA结合形成的核衣壳, 为prM蛋白和E蛋白提供锚定平台, 并参与病毒的侵入和组装过程[2]。NS1是一种具有2个保守N-连接糖基化位点和12个保守半胱氨酸的糖蛋白, 对病毒的复制至关重要。NS2A、NS2B、NS4A和NS4B是分子量较小的、疏水性的非结构蛋白。NS2B是NS3的辅助因子, 与NS3蛋白N端丝氨酸蛋白酶域在内质网膜上形成异源二聚体, 组成NS2B/NS3蛋白酶, 在病毒多聚蛋白切割过程中发挥着重要作用。NS3和NS5蛋白主要参与西尼罗病毒RNA复制的过程。NS3蛋白的C端为RNA解旋酶(helicase)域和核苷三磷酸酶(NTPase)域。Helicase和NTPase可以将复制产生的子代RNA链从模板链中分离, 与此同时, NTPase还能水解多核苷酸。NS5是最大的非结构蛋白, 其序列比较保守。它的N端为甲基转移酶(MTase)域, 对西尼罗病毒帽子进行甲基化, 增加其稳定性, C端为RNA依赖的RNA聚合酶(RdRp)域, RdRp以从头合成且不依赖引物的方式启动基因组RNA的合成[3]。

|
Figure 1 West Nile virus (WNV) particle illustration |

|
Figure 2 West Nile virus genome |
病毒感染敏感细胞后, 在其内进行自我复制(图 3)。复制环节主要分为: ①病毒粒子吸附于易感细胞表面; ②病毒E蛋白与细胞表面受体结合介导病毒侵入细胞, 若以E蛋白为靶点进行抑制, 则可阻止病毒的侵入。因此, E蛋白是研发西尼罗病毒抑制剂的重要靶点; ③病毒脂质包膜与细胞膜融合; ④病毒粒子脱壳和释放病毒RNA至细胞质中; ⑤病毒RNA翻译出一个多聚蛋白, 在NS2B/NS3蛋白酶和宿主蛋白酶的共同作用下将其切割为结构和非结构蛋白, NS2B/NS3蛋白酶和宿主蛋白酶在此步骤中不可或缺, 抑制它们可直接阻止病毒复制。因此, 是研发西尼罗病毒抑制剂的热门靶点; ⑥在RdRp和MTase作用下病毒RNA进行复制产生子代RNA, 若对它们进行靶向抑制, 则RNA复制无法完成; ⑦病毒RNA复制完成后, 在Helicase和NTPase作用下子代RNA从模板链中分离, 然后子代RNA再进行复制, 当它们受到抑制后子代RNA不能从模板链中分离, 后续的子代RNA复制将不能顺利进行; ⑧ C蛋白和子代RNA形成核衣壳后, 在内质网膜上与prM蛋白和E蛋白进行组装, 形成未成熟的病毒颗粒; ⑨未成熟的病毒颗粒沿着分泌途径穿过高尔基体, 在高尔基体中由弗林蛋白酶将prM蛋白裂解为M蛋白, 以促进病毒颗粒的成熟; ⑩成熟的病毒颗粒从感染细胞中释放出来[4]。完成整个复制周期, 进而感染新的宿主细胞。

|
Figure 3 West Nile virus life cycle. (Figure derived from Suthar MS, et al. Nat Rev Microbiol, 2013, 11:115-128) |
研究表明, 21%的患者在感染西尼罗病毒后会导致西尼罗热, 只有1%的患者会进一步恶化并发展为神经侵袭性疾病, 引起西尼罗病毒性脑炎。疾病的潜伏期通常为2~14天, 但在个别免疫功能低下的患者中发现潜伏期长达21天[5]。西尼罗热通常会突然发作, 临床表现为头痛、眼痛、肌痛、发烧、发冷、疲劳、呕吐和皮疹, 症状一般会持续数周或数月。皮疹通常出现在退烧时期, 表现为麻疹状、斑丘状和非瘙痒状, 主要在躯干和四肢上。西尼罗病毒脑炎与其他病毒性脑炎症状相似, 临床表现为严重的头痛和发烧, 伴有脑膜炎和畏光, 病情严重的患者会出现昏迷、呼吸困难直至呼吸循环衰竭导致死亡[6]。
对于无并发症的西尼罗热或西尼罗病毒性脑炎, 经过适当的治疗从而实现完全治愈。
2 抗西尼罗病毒抑制剂研究新进展正如上文所提到的, E蛋白、NS2B/NS3蛋白酶、NS5-MTase/NS5-RdRp、NTPase/helicase、宿主蛋白等已经成为目前抗西尼罗病毒属病毒抑制剂研究的主要靶点。
2.1 包膜蛋白(E蛋白)抑制剂包膜蛋白(E蛋白)是西尼罗病毒粒子表面的糖蛋白, 并且是病毒的抗原呈递蛋白。西尼罗病毒通过E蛋白介导和宿主细胞膜融合, 使病毒侵入宿主细胞。因此抑制E蛋白, 阻止病毒与宿主细胞的膜融合, 从而有效抑制西尼罗病毒的侵入[7]。
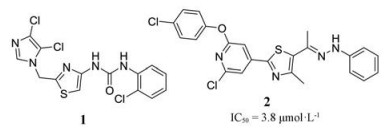
2009年, Kampmann等[8]基于登革热病毒(DEN) E蛋白结构进行化合物库的虚拟筛选, 从近135万个化合物中发现噻唑类登革热病毒抑制剂1、2。通过受感染细胞的膜融合实验, 证明了1、2能够抑制E蛋白介导的膜融合, 其IC50分别为32.0和1.2 μmol·L-1。并且2还可以抑制西尼罗病毒和黄热病毒E蛋白, 其IC50分别为3.8和1.6 μmol·L-1。
2.2 NS2B/NS3蛋白酶抑制剂蛋白酶抑制剂是一类治疗HIV感染的重要药物。丝氨酸蛋白酶抑制剂是治疗丙型肝炎病毒感染、糖尿病和心血管疾病的药物[9]。同样, NS3蛋白酶是一种丝氨酸蛋白酶, 含有经典的丝氨酸蛋白酶催化三联体, 由残基His51、Asp75和Ser135组成。只有当其与辅助因子NS2B结合形成异源二聚体(NS2B/NS3)才具有活性, 它在病毒多聚蛋白的裂解过程中发挥关键作用[10]。它是目前西尼罗病毒抑制剂研发的重要靶点, 现有的西尼罗病毒NS2B/NS3蛋白酶抑制剂主要分为肽类与非肽类。
|
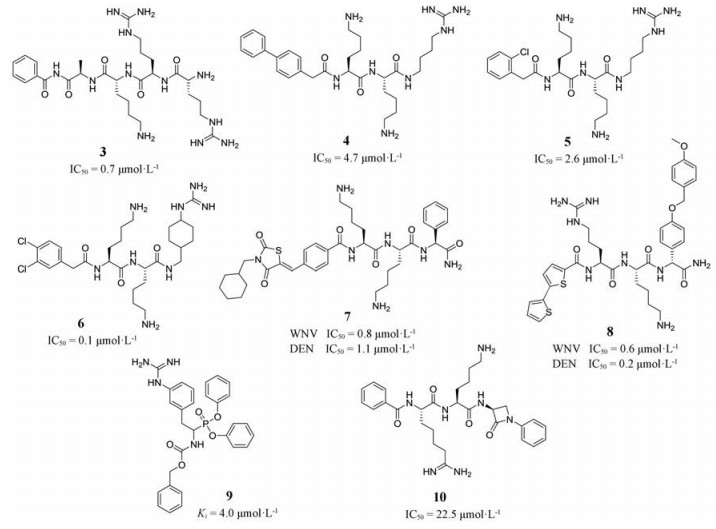
2006年, Knox等[11]设计合成一类多肽类抑制剂, 在体外抑制西尼罗病毒NS2B/NS3蛋白酶活性评价中发现代表抑制剂为3, 其IC50为0.7 μmol·L-1。
底物活性口袋位置是设计底物多肽类抑制剂最好的切入点, 将底物多肽添加上亲电性弹头可以使其转变为高效的靶向抑制剂。较常见的弹头是醛基, 例如, 含有醛基的肽类化合物能在亚微摩尔浓度下抑制西尼罗NS2B/NS3蛋白酶。然而由于化学稳定性差与选择性低, 限制其进一步开发。2011年, Lim等[12]通过模拟肽类底物设计了一类含有胍基丁胺基的肽类抑制剂, 它们不与活性位点上的丝氨酸共价结合, 而是通过疏水作用或静电作用与底物竞争活性位点。其中4是较强的西尼罗病毒NS2B/NS3蛋白酶抑制剂, 其IC50为4.7 μmol·L-1, 且稳定性好、易于合成, 值得作为先导化合物进行优化。2013年, 该研究小组报道了37种新型二肽类抑制剂, 代表抑制剂为5, 其IC50值为2.6 μmol·L-1 [13]。
2013年, Hammamy等[14]通过设计底物多肽类似物, 发现含有反式(4-胍)环己基甲胺(GCMA)基的肽类抑制剂与结合位点的适配性较好, 代表抑制剂为6, 其IC50为0.1 μmol·L-1。由于其活性好且稳定性高, 对胰蛋白酶类似物如纤维蛋白酶、Xa因子和基质酶无亲和力, 是一种比较有前景的抑制剂。
2013年, Nitsche等[15]通过设计底物多肽类似物, 发现一类含有噻唑烷二酮的登革热病毒NS2B/NS3蛋白酶抑制剂。基于此类抑制剂, 该课题组于2015年利用分子模拟技术对其进行结构优化, 得到了一种新型多肽类西尼罗病毒NS2B/NS3蛋白酶抑制剂7, 其IC50为0.8 μmol·L-1, 并能抑制登革热病毒NS2B/NS3蛋白酶, 其IC50为1.1 μmol·L-1。此研究初步证实了研发西尼罗病毒和登革热病毒NS2B/NS3蛋白酶双重抑制剂的可行性[16]。
2015年, Behnam等[17]对一类含有苯甘氨酸的底物多肽片段的C端和N端进行修饰。构效关系研究表明, 含有苄氧苯基甘氨酸的肽类抑制剂8能够显著抑制西尼罗病毒和登革热病毒NS2B/NS3蛋白酶, 其IC50分别为0.6和0.2 μmol·L-1, 其EC50分别为15.6和3.4 μmol·L-1, CC50均大于100.0 μmol·L-1, 并且具有很好的细胞膜透过性和代谢稳定性。
|
2018年, Skorenski等[18]发现一类含有二苯磷酸的肽类西尼罗病毒NS2B/NS3蛋白酶抑制剂, 代表抑制剂为9, 其抑制常数(Ki)为4.0 μmol·L-1。9是一种不可逆抑制剂, 通过与活性位点丝氨酸残基发生特异性结合, 从而形成蛋白酶抑制剂复合物而发挥作用。
最近, Drazic等[19]合成一类含有亲电性“弹头”β-内酰胺环的肽类西尼罗病毒和登革热病毒NS2B/NS3蛋白酶双重抑制剂, 发现10对西尼罗病毒NS2B/NS3蛋白酶抑制作用最强, 其IC50值为22.5 μmol·L-1, 且能够抑制登革热病毒NS2B/NS3蛋白酶。10通过共价键与NS2B/NS3蛋白酶结合, 表现出较低的脱靶效应, 对凝血酶和胰蛋白酶几乎无亲和力。
构效研究发现, 活性较好的肽类抑制剂中都存在赖氨酸或精氨酸残基, 从而推测赖氨酸或精氨酸残基有利于提高多肽类抑制剂活性, 并且多肽的C端和N端均是结构修饰的有利位点。
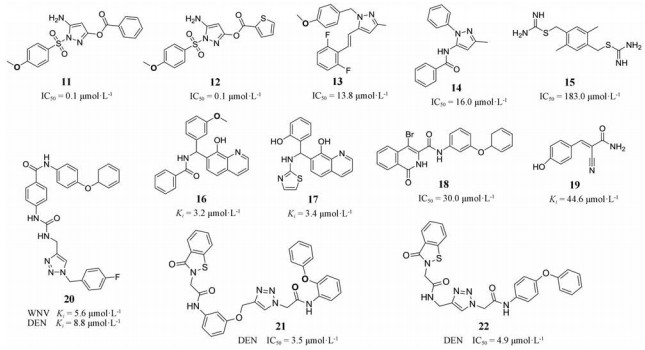
2.2.2 非肽类抑制剂2007年, Johnston等[20]通过对65 000个化合物进行高通量筛选, 发现2个西尼罗病毒NS2B/NS3蛋白酶抑剂11和12, 其IC50均为0.1 μmol·L-1。然而, 在pH为8的缓冲液中11和12被快速水解为吡唑-3-醇。因此, 为了提高它们的化学稳定性, 通过结构改造得到两种非酯类抑制剂13和14, 其IC50分别为13.8和16.0 μmol·L-1, 尽管它们的活性降低, 但其在pH为8的缓冲液中的降解时间延长至13 h和96 h, 稳定性大幅提高, 是一类比较有前景的抑制剂[21]。
2008年, Ekonomiuk等[22]通过对近12 000个化合物进行高通量筛选, 发现一种硫脲类西尼罗病毒NS2B/NS3蛋白酶抑制剂15, 其IC50为183.0 μmol·L-1。由于15相对分子质量较小, 只占据底物结合位点的两个口袋S1和S2 (图 4), 仍有结构修饰的空间, 分子模拟表明, 15的苯环上一个或两个甲基取代会提高其活性, 因此可作为先导化合物供进一步优化。

|
Figure 4 Molecular docking of compound 15 onto WNV NS2B/NS3 protease. (The red dotted line represents hydrogen bonds) |
2008年, Mueller等[23]通过对近31 000个化合物进行高通量筛选, 发现98个西尼罗病毒NS2B/NS3蛋白酶抑制剂, 其中8-羟基喹啉类化合物16和17为代表抑制剂, 其Ki分别为3.2和3.4 μmol·L-1。值得注意的是, 17有一个手性中心, 分子模拟表明其不同的构象会影响与蛋白酶的结合力。17的R构象与蛋白酶形成三重氢键, 其中8-羟基喹啉基团中的羟基和氮原子分别与S4口袋形成两重氢键, 主链上的氮原子和S2口袋形成第三重氢键。与R构象形式相比, 17的S构象只与蛋白酶形成两重氢键(图 5)。进一步的构效关系研究表明8-羟基喹啉是活性必需基团[24]。
|

|
Figure 5 Molecular docking of compound 17 (R and S enantiomer) onto WNV NS2B/NS3 protease (The red dotted line represents hydrogen bonds) |
2010年, Dou等[25]以异喹啉为骨架, 设计合成了一类西尼罗病毒NS2B/NS3蛋白酶抑制剂。代表抑制剂为18, 其IC50为30.0 μmol·L-1, 选择性较好, 对哺乳动物丝氨酸蛋白酶无亲和力。
2011年, Nitsche等[26]通过将苯丙烯基与亲电试剂腈基结合, 合成了86个氰基丙烯酰胺类NS3/NS2B蛋白酶抑制剂, 代表抑制剂为19, 其Ki为44.6 μmol·L-1。构效关系研究表明芳香基团和中心双键是其活性的关键。
2012年, Aravapalli等[27]以氨基苯甲酰胺为骨架, 通过点击化学(click chemistry)合成了一类西尼罗病毒NS3/NS2B蛋白酶抑制剂, 代表抑制剂为20, 其Ki为5.6 μmol·L-1。此外, 20对登革热病毒NS3/NS2B蛋白酶也具有亲和力, 其Ki为8.8 μmol·L-1。
2012年, Tiew等[28]通过点击化学设计合成了一类对登革热病毒和西尼罗病毒NS2B/NS3蛋白酶均有活性的三唑类抑制剂。其中21和22抑制登革热病毒NS2B/NS3蛋白酶的IC50分别为3.5和4.9 μmol·L-1, 它们对西尼罗病毒NS2B/NS3蛋白酶抑制作用相对较弱。
通过分析非肽类抑制剂的结构特点, 发现活性较好的抑制剂中普遍存在芳香环和亲电子基团, 推测芳香环有利于提高抑制剂的活性, 亲电子基团可以增强活性。
通过对两种不同类型的抑制剂进行总结(表 1), 发现肽类抑制剂相对分子质量较大, 稳定性与选择性低于非肽类抑制剂, 可以通过降低分子尺寸进行结构优化, 以提高活性。非肽类抑制剂相对分子质量比肽类的小且选择性与稳定性更高, 但是活性普遍低于肽类抑制剂, 可以通过增加或降低分子尺寸进行结构优化, 以提高活性。
| Table 1 The differences between peptides and non-peptides |
NS5是西尼罗病毒最大的非结构蛋白, 其N端为MTase域, C端为RdRp域, 当其受到抑制时, 病毒复制是不能完成的[29]。
2009年, Puig-Basagoiti等[30]对近10万个化合物进行高通量筛选, 发现5个西尼罗病毒抑制剂23~27 (图 6), 随后对多种黄病毒和非黄病毒进行活性筛选, 发现这5个化合物具有不同的抗病毒特性。其中23和24能够同时抑制西尼罗病毒、甲病毒和杆状病毒, 它们抑制西尼罗病毒的EC50分别为9.0和2.0 μmol·L-1, CC50分别为125.0和45.0 μmol·L-1。25能够抑制西尼罗病毒和登革热病毒, 其EC50分别为7.0和8.0 μmol·L-1, CC50分别为77.0和80.0 μmol·L-1。26能够抑制3种黄病毒(西尼罗病毒、登革热病毒和黄热病毒), 其EC50分别为0.7、1.5和1.5 μmol·L-1, CC50分别为14.7、15.0和15.0 μmol·L-1, 但不抑制非黄病毒。27只抑制西尼罗病毒, 其EC50为4.0 μmol·L-1, CC50为52.0 μmol·L-1。

|
Figure 6 The chemical structures of NS5-MTase/NS5-RdRp inhibitors 23-30 |
胸苷类化合物28 (图 6)是一种强效的HIV-1抑制剂。构效关系研究表明, 三唑5位较大的基团是活性的关键[31]。29 (图 6)是一种结合模式独特的HIV非核苷逆转录酶抑制剂, 研究者根据这两类抑制剂的结构特点, 将28中四氢呋喃环的5位引入甲硅烷基合成一种新型胸苷类化合物30 (图 6)。意外的是, 30没有抑制HIV-1活性。与此同时, 在对其他病毒进行活性筛选时, 发现30是西尼罗病毒和登革热病毒选择性抑制剂, 其EC50分别为2.9和7.3 μmol·L-1, CC50分别为13.0和7.9 μmol·L-1; 对流感病毒、人类巨细胞病毒或丙型肝炎病毒均无抑制作用[32]。
2.4 NTPase/helicase抑制剂西尼罗病毒NTPase/helicase可以将复制产生的子代RNA链从模板链中分离, 并且NTPase能够水解多核苷酸[33]。因此, 抑制NTPase/helicase可以直接阻止西尼罗病毒的复制。
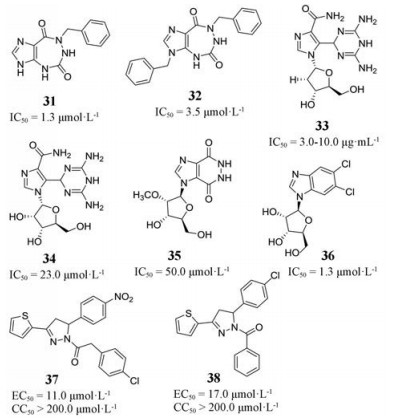
2003年, Zhang等[34]设计合成了一类扩环的核苷酸类西尼罗病毒NTPase/helicase抑制剂, 同时也测试了其对人Suv3蛋白酶的抑制活性。其中代表性化合物为31和32, 对NTPase/helicase的抑制活性较好, 其IC50值分别为1.3和3.5 μmol·L-1, 而对Suv3蛋白酶几乎没有抑制活性(IC50 > 500.0 μmol·L-1), 预示此类化合物对宿主毒性可能较低。
2005年, Ujjinamatada等[35]从一类具有潜在抗病毒和抗癌活性的化合物库中发现一种西尼罗病毒NTPase/helicase抑制剂33, 在使用RNA底物的活性测试时, 其IC50值为3.0~10.0 μg·mL-1。然而, 在使用DNA底物的活性测试中, 却无抑制作用。有趣的是, 33的类似物34, 在使用DNA底物的活性测试中, 却对西尼罗病毒NTPase/helicase有抑制活性, 其IC50值23.0 μmol·L-1, 但是在使用RNA底物重复同样的实验时, 却无抑制活性[36]。类核苷抑制剂35和36同样具有抑制西尼罗病毒NTPase/helicase活性, 其IC50值分别为50.0和3.0 μmol·L-1 [37, 38]。
2006年, Goodell等[39]对108个具有抗菌和抗病毒活性的化合物进行高通量筛选, 发现了2个吡唑啉类西尼罗病毒抑制剂37和38, 在BHK细胞(EC50分别为11.0和17.0 μmol·L-1, CC50均大于200.0 μmol·L-1)和Vero细胞(EC50分别为25.0和23.0 μmol·L-1, CC50均大于200.0 μmol·L-1)中均显示出良好的抗西尼罗病毒活性。
|
DDX3X是一种宿主蛋白, 近年来的研究发现, 它能够参与西尼罗病毒复制过程中转录、翻译和RNA降解等过程。因此, 抑制DDX3X可以有效阻止西尼罗病毒的复制。
Brai等[40]发现第一个广谱抗病毒DDX3X抑制剂39, 具有良好的抑制西尼罗病毒活性, 其EC50为8.8 μmol·L-1, CC50大于200.0 μmol·L-1。但其水溶性差, 容易在脂肪组织中积累导致生物利用度较低。为了改善其水溶性, 通过基于同源建模的虚拟筛选发现了苯磺酰胺类抑制剂40, 其对西尼罗病毒抑制活性有所降低(EC50为120.0 μmol·L-1, CC50为175.0 μmol·L-1), 但其水溶性增强, 生物利用度有所提高。在构效关系分析的基础上, 将39中三唑与40中的苯磺酰胺进行“分子杂合”设计出一种新型抑制剂41 (图 7), 其EC50为2.3 μmol·L-1, CC50大于200.0 μmol·L-1, 由于活性较好且细胞毒性低, 是一类前景较好的抑制剂。

|
Figure 7 The chemical structures of host-targeted inhibitors 39-41 |
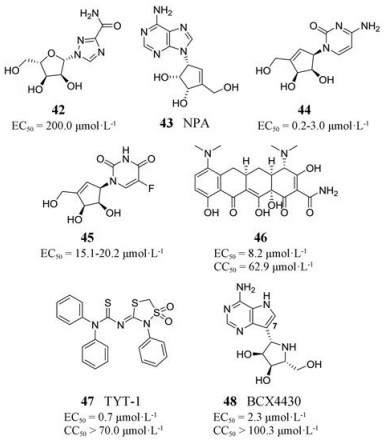
Morrey等[41]通过细胞培养和啮齿类动物模型实验发现广谱抗病毒药物利巴韦林42在体外具有微弱的抗西尼罗病毒活性(EC50约200.0 μmol·L-1), 但在动物体内毒性较大。研究发现, 利用利巴韦林治疗感染西尼罗病毒的叙利亚金仓鼠, 反而会增加其死亡率。
NPA (43)是一种具有抗肿瘤和抗病毒活性的腺苷类似物, 由于细胞毒性较大, 使其进一步研究受限。Song等[42]通过合成NPA类似物, 发现胞嘧啶类似物44和5-氟胞嘧啶类似物45在体外具有较强抗西尼罗病毒活性, 其EC50分别为0.2~3.0和15.1~20.2 μmol·L-1。
米诺环素(46)是一种临床常用、耐受性良好的抗生素, 作为一种治疗西尼罗病毒感染的药物, 它在治疗西尼罗病毒性脑炎过程中显示出优良的神经保护、抗病毒和抗炎作用。2007年, Michaelis等[43]验证米诺环素在Vero细胞中具有明显的抗西尼罗病毒活性, 其EC50约为8.2 μmol·L-1, CC50约为62.9 μmol·L-1。
2007年, Barklis等[44]通过对3 500个化合物进行高通量筛选, 发现4个硫脲类西尼罗病毒抑制剂, 代表抑制剂为TYT-1 (47), 其EC50为0.7 μmol·L-1, CC50大于70.0 μmol·L-1。进一步研究发现该抑制剂能够阻止西尼罗病毒复制的翻译过程, 然而, 其确切机制仍不清楚。
|
BCX4430 (48)是一种广谱抗病毒抑制剂。它在核糖环上以氮代氧, 在杂碱基部分的7位上以碳代氮, 是亚氨基-C核苷的典型代表。BCX4430使依赖于tRNA的RNA聚合酶受到抑制, 导致病毒RNA子代链复制的终止。Eyer等[45]研究发现, BCX4430在体外能强烈抑制西尼罗病毒复制(EC50 = 2.3 μmol·L-1), 且细胞毒性较小(CC50 > 100.3 μmol·L-1)。
通过对不同作用靶点的代表抑制剂活性进行总结(表 2), 发现NS2B/NS3蛋白酶抑制剂6在所有抑制剂中活性最好, 作为肽类抑制剂, 其稳定性和选择性相比其他肽类抑制剂得到一定提升。NS5-MTase/NS5-RdRp抑制剂26虽然活性与6相差不是很大, 但是其细胞毒性较大, 限制了其进一步研发。NTPase/helicase抑制剂31的活性虽然比6的差, 但是其选择性更好, 对宿主细胞Suv3蛋白酶几乎无抑制作用。宿主蛋白抑制剂41虽然活性相比6略有不足, 但是其稳定性更好, 且细胞毒性较小。
| 表 2 Activity of representative inhibitors with different targets |
西尼罗病毒正在全球迅速传播, 由于其感染可能导致严重的病毒性脑炎, 已成为威胁人类健康的一类恶性传染病。近年来, 尽管研究人员针对西尼罗病毒各靶点进行大量的研究, 但是仍没有特异性抗西尼罗病毒药物被批准上市。并且现有的西尼罗病毒抑制剂由于活性问题、安全性低、以及药代动力学性质不佳等不足, 尚不能满足治愈西尼罗病毒感染的要求。虽然目前我国尚未发现西尼罗病毒感染病例, 为了防止其在我国的突然爆发流行, 尽早研发出有效的防治西尼罗病毒药物, 对保障国民健康具有重要意义。
众所周知, 药物研发周期长、投入多、风险高。近年来, 由于研发成本不断增加、重磅药物专利到期后导致其竞争力下降、创新药物药效提升不明显以及越来越严格的监管政策, 导致创新药物研发的投入与产出比例严重失衡, 导致科研人员对研发新型西尼罗病毒抑制剂的兴趣降低。因此新策略以及新方法的应用在新型西尼罗病毒抑制剂研发过程中就显得尤为重要[46]。
老药新用是解决上述问题、提高药物发现效率的一种重要方法。老药新用, 是指已批准上市或者未上市但结构明确、生物活性已知的药物, 通过进一步研究发现其新的作用靶点以扩大其适应症范围。上市药物经过长期临床实践, 其毒副作用均已明确, 且生产工艺、质量标准、剂型等完备。可以通过对经典已上市或未上市但活性较好的抗病毒药物进行抗西尼罗病毒活性筛选, 发现新的优势化合物, 从而进行再优化或直接使用, 以缩短研发时间和降低研发成本, 提高研发的成功率, 有效抑制西尼罗病毒的传播。除此之外, 通过底物多肽类似物进行更为合理及精准的药物设计, 以期发现细胞活性更好, 具有更好药代动力学性质的抑制剂。在今后的研发中, 随着计算化学和人工智能的广泛应用, 科研人员可以进行更为精准的虚拟筛选, 从而提高研发成功率。相信随着研究的不断深入, 越来越多的高效抑制剂将会问世。
| [1] |
Saxena V, Bolling BG, Wang T. West Nile virus[J]. Clin Lab Med, 2017, 37: 243-252. DOI:10.1016/j.cll.2017.01.001 |
| [2] |
Kramer LD, Li J, Shi PY. West Nile virus[J]. Lancet Neurol, 2007, 6: 171-181. DOI:10.1016/S1474-4422(07)70030-3 |
| [3] |
Brinton MA. The molecular biology of West Nile virus: a new invader of the western hemisphere[J]. Annu Rev Microbiol, 2002, 56: 371-402. DOI:10.1146/annurev.micro.56.012302.160654 |
| [4] |
Suthar MS, Diamond MS, Gale M. West Nile virus infection and immunity[J]. Nat Rev Microbiol, 2013, 11: 115-128. DOI:10.1038/nrmicro2950 |
| [5] |
Zou S, Foster GA, Dodd RY, et al. West Nile fever characteristics among viremicpersons identified through blood donor screening[J]. J Infect Dis, 2010, 202: 1354-1361. DOI:10.1086/656602 |
| [6] |
Petersen LR, Brault AC, Nasci RS. West Nile virus: review of the literature[J]. JAMA, 2013, 310: 308-315. DOI:10.1001/jama.2013.8042 |
| [7] |
Deng YQ, Dai JX, Ji GH, et al. A broadly flavivirus cross-neutralizing monoclonal antibody that recognizes a novel epitope within the fusion loop of E protein[J]. PLoS One, 2011, 6: e16059. DOI:10.1371/journal.pone.0016059 |
| [8] |
Kampmann T, Yennamalli R, Campbell P, et al. In silico screening of small molecule libraries using the Dengue virus envelope E protein has identified compounds with antiviral activity against multiple flaviviruses[J]. Antiviral Res, 2009, 84: 234-241. DOI:10.1016/j.antiviral.2009.09.007 |
| [9] |
Stoermer MJ, Chappell KJ, Liebscher S, et al. Potent cationic inhibitors of West Nile virus NS2B/NS3 protease with serum stability, cell permeability and antiviral activity[J]. J Med Chem, 2008, 51: 5714-5721. DOI:10.1021/jm800503y |
| [10] |
Pinkham AM, Yu Z, Cowan JA. Attenuation of West Nile virus NS2B/NS3 protease by amino terminal copper and nickel binding (ATCUN) peptides[J]. J Med Chem, 2018, 61: 980-988. DOI:10.1021/acs.jmedchem.7b01409 |
| [11] |
Knox JE, Ma NL, Yin Z, et al. Peptide inhibitors of West Nile NS3 protease: SAR study of tetrapeptide aldehyde inhibitors[J]. J Med Chem, 2006, 49: 6585-6590. DOI:10.1021/jm0607606 |
| [12] |
Lim HA, Joy J, Hill J, et al. Novel agmatine and agmatine-like peptidomimetic inhibitors of the West Nile virus NS2B/NS3 serine protease[J]. Eur J Med Chem, 2011, 46: 3130-3134. DOI:10.1016/j.ejmech.2011.04.055 |
| [13] |
Lim HA, Ang MJ, Joy J, et al. Novel agmatine dipeptide inhibitors against the West Nile virus NS2B/NS3 protease: a P3 and N-cap optimization study[J]. Eur J Med Chem, 2013, 62: 199-205. DOI:10.1016/j.ejmech.2012.12.043 |
| [14] |
Hammamy MZ, Haase C, Hammami M, et al. Development and characterization of new peptidomimetic inhibitors of the West Nile virus NS2B-NS3 protease[J]. ChemMedChem, 2013, 8: 231-241. DOI:10.1002/cmdc.201200497 |
| [15] |
Nitsche C, Schreier VN, Behnam MA, et al. Thiazolidinone-peptide hybrids as Dengue virus protease inhibitors with antiviral activity in cell culture[J]. J Med Chem, 2013, 56: 8389-8403. DOI:10.1021/jm400828u |
| [16] |
Bastos Lima A, Behnam MA, El Sherif Y, et al. Dual inhibitors of the Dengue and West Nile virus NS2B-NS3 proteases: synthesis, biological evaluation and docking studies of novel peptide-hybrids[J]. Bioorg Med Chem, 2015, 23: 5748-5755. DOI:10.1016/j.bmc.2015.07.012 |
| [17] |
Behnam MA, Graf D, Bartenschlager R, et al. Discovery of nanomolar Dengue and West Nile virus protease inhibitors containing a 4-benzyloxyphenylglycine residue[J]. J Med Chem, 2015, 58: 9354-9370. DOI:10.1021/acs.jmedchem.5b01441 |
| [18] |
Skoreński M, Milewska A, Pyrć K, et al. Phosphonate inhibitors of West Nile virus NS2B/NS3 protease[J]. J Enzyme Inhib Med Chem, 2019, 34: 8-14. DOI:10.1080/14756366.2018.1506772 |
| [19] |
Dražić T, Kopf S, Corridan JF, et al. Peptide-β-lactam inhibitors of Dengue and West Nile virus NS2B-NS3 protease display two distinct binding modes[J]. J Med Chem, 2020, 63: 140-156. DOI:10.1021/acs.jmedchem.9b00759 |
| [20] |
Johnston PA, Phillips J, Shun TY, et al. HTS identifies novel and specific uncompetitive inhibitors of the two-component NS2B-NS3 proteinase of West Nile virus[J]. Assay Drug Dev Technol, 2007, 5: 737-750. DOI:10.1089/adt.2007.101 |
| [21] |
Sidique S, Shiryaev SA, Ratnikov BI, et al. Structure-activity relationship and improved hydrolytic stability of pyrazole derivatives that are allosteric inhibitors of West Nile virus NS2B-NS3 proteinase[J]. Bioorg Med Chem Lett, 2009, 19: 5773-5777. DOI:10.1016/j.bmcl.2009.07.150 |
| [22] |
Ekonomiuk D, Su XC, Ozawa K, et al. Discovery of a non-peptidic inhibitor of West Nile virus NS3 proteaseby high-throughput docking[J]. PLoS Negl Trop Dis, 2009, 3: e356. DOI:10.1371/journal.pntd.0000356 |
| [23] |
Mueller NH, Pattabiraman N, Ansarah-Sobrinho C, et al. Identification and biochemical characterization of small-molecule inhibitors of West Nile virus serine protease by a high-throughput screen[J]. Antimicrob Agents Chem, 2008, 52: 3385-3393. DOI:10.1128/AAC.01508-07 |
| [24] |
Ezgimen M, Lai H, Mueller NH, et al. Characterization of the 8-hydroxyquinoline scaffold for inhibitors of West Nile virus serine protease[J]. Antiviral Res, 2012, 94: 18-24. DOI:10.1016/j.antiviral.2012.02.003 |
| [25] |
Dou D, Viwanathan P, Li Y, et al. Design, synthesis, and in vitro evaluation of potential West Nile virus protease inhibitors based on the 1-oxo-1, 2, 3, 4-tetrahydroisoquinoline and 1-oxo-1, 2-dihydroisoquinoline scaffolds[J]. J Comb Chem, 2010, 12: 836-843. DOI:10.1021/cc100091h |
| [26] |
Nitsche C, Steuer C, Klein CD. Arylcyanoacrylamides as inhibitors of the Dengue and West Nile virus proteases[J]. Bioorg Med Chem, 2011, 19: 7318-7337. DOI:10.1016/j.bmc.2011.10.061 |
| [27] |
Aravapalli S, Lai H, Teramoto T, et al. Inhibitors of Dengue virus and West Nile virus proteases based on the aminobenzamide scaffold[J]. Bioorg Med Chem, 2012, 20: 4140-4148. DOI:10.1016/j.bmc.2012.04.055 |
| [28] |
Tiew KC, Dou D, Teramoto T, et al. Inhibition of Dengue virus and West Nile virus proteases by click chemistry-derived benz[d]isothiazol-3(2H)-one derivatives[J]. Bioorg Med Chem, 2012, 20: 1213-1221. |
| [29] |
Lopez-Denman AJ, Russo A, Wagstaff KM, et al. Nucleocytoplasmic shuttling of the West Nile virus RNA-dependent RNA polymerase NS5 is critical to infection[J]. Cell Microbiol, 2018, 20: e12848. |
| [30] |
Puig-Basagoiti F, Qing M, Yuan Z, et al. Identification and characterization of inhibitors of West Nile virus[J]. Antiviral Res, 2009, 83: 71-79. DOI:10.1016/j.antiviral.2009.03.005 |
| [31] |
Sirivolu VR, Vernekar SKV, Ilina T, et al. Clicking 3'-azidothymidine into novel potent inhibitors of human immunodeficiency virus[J]. J Med Chem, 2013, 56: 8765-8780. DOI:10.1021/jm401232v |
| [32] |
Vernekar SK, Qiu L, Zhang J, et al. 5'-Silylated 3'-1, 2, 3-triazolyl thymidine analogues as inhibitors of West Nile virus and Dengue virus[J]. J Med Chem, 2015, 58: 4016-4028. DOI:10.1021/acs.jmedchem.5b00327 |
| [33] |
Setoh YX, Periasamy P, Peng NYG, et al. Helicase domain of West Nile virus NS3 protein plays a role in inhibition of type I interferon signaling[J]. Viruses, 2017, 9: 326. DOI:10.3390/v9110326 |
| [34] |
Zhang N, Chen HM, Koch V, et al. Ring-expanded ("fat") nucleoside and nucleotide analogues exhibit potent in vitro activity against Flaviviridae NTPases/helicases, including those of the West Nile virus, hepatitis C virus, and Japanese Encephalitis virus[J]. J Med Chem, 2003, 46: 4149-4164. DOI:10.1021/jm030842j |
| [35] |
Ujjinamatada RK, Agasimundin YS, Zhang P, et al. A novel imidazole nucleoside containing a diaminodihydro-S-triazine as a substituent: inhibitory activity against the West Nile virus NTPase/helicase[J]. Nucleosides Nucleotides Nucleic Acids, 2005, 24: 1775-1788. DOI:10.1080/15257770500267063 |
| [36] |
Ujjinamatada RK, Baier A, Borowski P, et al. An analogue of AICAR with dual inhibitory activity against WNV and HCV NTPase/helicase: synthesis and in vitro screening of 4-carbamoyl-5-(4, 6-diamino-2, 5-dihydro-1, 3, 5-triazin-2-yl)imidazole-1-beta-D-ribofuranoside[J]. Bioorg Med Chem Lett, 2007, 17: 2285-2288. DOI:10.1016/j.bmcl.2007.01.074 |
| [37] |
Borowski P, Lang M, Haag A, et al. Characterization of imidazo[4, 5-d]pyridazine nucleosides as modulators of unwinding reaction mediated by West Nile virus nucleoside triphosphatase/helicase: evidence for activity on the level of substrate and/or enzyme[J]. Antimicrob Agents Chemother, 2002, 46: 1231-1239. DOI:10.1128/AAC.46.5.1231-1239.2002 |
| [38] |
Borowski P, Deinert J, Schalinski S, et al. Halogenated benzimidazoles and benzotriazoles as inhibitors of the NTPase/helicase activities of hepatitis C and related viruses[J]. Eur J Biochem, 2003, 270: 1645-1653. DOI:10.1046/j.1432-1033.2003.03540.x |
| [39] |
Goodell JR, Puig-Basagoiti F, Forshey BM, et al. Identification of compounds with anti-West Nile virus activity[J]. J Med Chem, 2006, 49: 2127-2137. DOI:10.1021/jm051229y |
| [40] |
Brai A, Martelli F, Riva V, et al. DDX3X helicase inhibitors as a new strategy to fight the West Nile virus infection[J]. J Med Chem, 2019, 62: 2333-2347. DOI:10.1021/acs.jmedchem.8b01403 |
| [41] |
Morrey JD, Day CW, Julander JG, et al. Effect of interferon-alpha and interferon-inducers on West Nile virus in mouse and hamster animal models[J]. Antivir Chem Chemother, 2004, 15: 101-109. |
| [42] |
Song GY, Paul V, Choo H, et al. Enantiomeric synthesis of D-and L-cyclopentenyl nucleosides and their antiviral activity against HIV and West Nile virus[J]. J Med Chem, 2001, 44: 3985-3993. DOI:10.1021/jm010256v |
| [43] |
Michaelis M, Kleinschmidt MC, Doerr HW, et al. Minocycline inhibits West Nile virus replication and apoptosis in human neuronal cells[J]. J Antimicrob Chemother, 2007, 60: 981-986. DOI:10.1093/jac/dkm307 |
| [44] |
Barklis E, Still A, Sabri MI, et al. Sultam thiourea inhibition of West Nile virus[J]. Antimicrob Agents Chemother, 2007, 51: 2642-2645. DOI:10.1128/AAC.00007-07 |
| [45] |
Eyer L, Zouharová D, Širmarová J, et al. Antiviral activity of the adenosine analogue BCX4430 against West Nile virus and tick-borne flaviviruses[J]. Antiviral Res, 2017, 142: 63-67. DOI:10.1016/j.antiviral.2017.03.012 |
| [46] |
Wu G, Zhao T, Kang D, et al. Overview of recent strategic advances in medicinal chemistry[J]. J Med Chem, 2019, 62: 9375-9414. DOI:10.1021/acs.jmedchem.9b00359 |