 2020, Vol. 55
2020, Vol. 55



痘病毒(poxvirus)是所有病毒中最大、最复杂的病毒, 感染人和动物后会造成局部或全身化脓性皮肤损害。与哺乳动物相关的可分为8个属:正痘病毒属(Orthopoxvirus, OPXV)、山羊痘病毒属、禽痘病毒属、副痘病毒属、猪痘病毒属、兔痘病毒属、软疣痘病毒属和亚塔痘病毒属。其中正痘病毒属、副痘病毒属、软疣病毒属和亚塔痘病毒属中的部分病毒具有感染人的能力(表 1)[1]。
| Table 1 Classification and features of poxviruses infecting humans[4] |
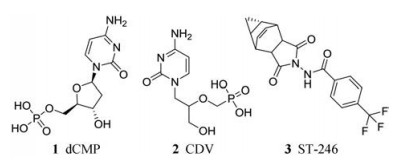
正痘病毒属是哺乳动物亚科中成员最多、宿主范围最广和危害最严重的一个属, 其中天花病毒(smallpox virus, VARV)会导致高致死率的烈性传染病—天花。1980年5月世界卫生组织宣布人类成功消灭天花, 并叫停了疫苗接种计划[2]。现如今未注射疫苗群体的免疫力已经下降, 另一方面猴痘、牛痘等人兽共患痘病毒病频繁出现, 恐怖主义也可能利用其进行生物袭击。这些潜在的危险催促着新药的研发, 用来预防天花以及其他痘病毒感染的暴发[3]。虽然疫苗接种在预防和控制天花病毒方面占有主要地位, 但对于不能接种疫苗和已感染的人来说药物治疗也是十分重要的。因此, 寻找能够有效对抗天花病毒及其他痘病毒的药物刻不容缓。西多福韦(cidofovir, CDV)和替韦立马(tecovirimat, ST-246)是目前已上市的抗痘病毒药物, 但依然存在着口服利用度、耐药性、肾毒性等问题。
随着对痘病毒结构和复制周期的深入了解, 发现其复制过程中的功能蛋白及其相关的基因产物都有可能成为药物开发的新靶点, 目前已经发现了许多潜在靶点及相关抑制剂, 有望发展成为新型抗痘病毒感染药物。
1 痘病毒的生物学 1.1 痘病毒的结构及基因组病毒粒呈砖形或椭圆形, 大小为(300~450) × (170~260) nm (图 1)。由一个双面凹的核心、两个侧体和包膜组成。病毒的基因组集中在核心内, 核心内有多种结构蛋白和酶。

|
Figure 1 Structural graphic of poxvirus |
痘病毒基因组为单分子的线性双股DNA (128~365 kb), 编码200多个基因。末端具有长倒置重复序列, 形成一个单股的环[5]。
1.2 痘病毒的复制周期概述痘病毒在复制过程中会产生两种感染性的病毒颗粒:细胞内成熟病毒(intracellular mature virus, IMV)和细胞外包膜病毒(extracellular enveloped virus, EEV)。痘病毒的复制周期可分为以下几个阶段:侵入和脱壳、早期转录、病毒DNA复制、晚期转录、病毒颗粒组装以及释放(图 2)[6]。

|
Figure 2 The life cycle of poxvirus replication. Extracellular enveloped virions (EEV), intracellular mature virions (IMV), immature virions (Ⅳ), intracellular enveloped virions (IEV), and cell associated enveloped virions (CEV) are depicted |
侵入和脱壳: IMV或EEV外膜上的病毒蛋白与宿主细胞上的受体结合, 吞饮入细胞内, 通过水解酶的酶解作用脱壳, 使病毒核心释放到细胞质中。
早期转录:病毒基因在DNA依赖的RNA聚合酶的作用下进行早期转录, 合成与DNA复制相关的酶以及免疫调节蛋白。
病毒DNA复制:病毒基因组在DNA聚合酶和其他因子的作用下进行复制。
晚期转录:病毒DNA复制完成后, 进行晚期转录, 生成结构蛋白以及早期组装所需的酶。
病毒颗粒的组装与释放:未成熟的病毒粒子(immature virions, Ⅳ)组装并加工成IMV。大多数IMV是通过细胞裂解从细胞中释放出来的, IMV颗粒被双层膜包裹形成细胞内的包膜病毒(intracellular enveloped virions, IEV), 然后通过微管运输到细胞表面, 外膜与细胞膜融合, 病毒释放为EEV。
2 已上市药物及其作用机制 2.1 西多福韦(cidofovir, CDV)CDV是最早用于天花暴发的应急治疗药物, 在临床上主要用于巨细胞病毒视网膜炎的治疗[7]。西多福韦属于无环磷酸核苷类化合物, 是一种单磷酸脱氧胞苷(dCMP)类似物。作为一个广谱的双链DNA病毒抑制剂, CDV对痘病毒、疱疹病毒以及腺病毒都有很好的抑制作用。CDV在临床使用过程中存在着口服生物利用度不足、只能静脉注射给药以及肾毒性等缺点。因此, 成药性是后续化合物结构优化的目标。CDV被感染细胞摄取后, 转化成磷酸盐并与感染细胞内的脱氧胞苷三磷酸(dCTP)竞争, 在DNA聚合酶水平阻碍病毒DNA的合成, 从而抑制病毒的复制[8]。
2.2 替韦立马(tecovirimat, ST-246)替韦立马是2018年7月13日被美国FDA获批上市的一类新型天花治疗药物, 与CDV相比, 口服利用度、药代动力学性质及药物毒性都有很大的改善[9]。
替韦立马靶向于痘病毒F13L, F13L编码产生细胞外病毒所需的主要包膜蛋白P37。替韦立马抑制P37的生成从而达到抑制细胞外病毒的生成、阻止病毒细胞间传播的目的[10]。
|
理论上, 在病毒的整个复制周期中, 任一环节都可以作为抑制剂的靶标。尽管近年来报道了大量具有抑制痘病毒活性的化合物, 但仍存在着活性低、细胞毒性大以及溶解性差等问题。因此研发新一代高效、低毒且作用机制明确的抗正痘病毒药物依然是药物化学工作者的重要任务。
正痘病毒的复制周期中涉及众多功能蛋白和基因产物, 且作用机制复杂。按照复制周期的阶段和靶点的不同, 各类抑制剂如下文所述。
3.1 侵入和脱壳抑制剂IMV和EEV颗粒与细胞表面结合从而侵入细胞, 其过程涉及H3L、A27L和D8L编码的蛋白与细胞糖胺聚糖的相互作用[11]。病毒核心的脱壳是一个复杂的过程, 涉及A16L、A21L、A28L、F9L、G3L、G9R、H2R、J5L和L5R这几种病毒蛋白[12-14]。
目前有效的侵入和脱壳抑制剂以各类单克隆抗体为主。研究证明, 针对A17L和L1R的单克隆抗体可以在早期抑制病毒感染, 而针对H3L和B5R的小鼠和人源单克隆抗体在牛痘病毒感染的动物模型中是有效的[15]。
3.2 DNA复制抑制剂DNA合成的启动涉及I1L、I6L和K4L这3种病毒蛋白[16]。然后在5种酶的参与下完成DNA合成, 即E9L DNA聚合酶、核苷三磷酸酶(D5R)、B1R蛋白激酶、尿嘧啶DNA糖基化酶(D4R)和聚合酶持续性因子(A20R)。其中A20R蛋白、D4R蛋白和DNA聚合酶一起构成全酶。D4R可以从病毒DNA中切除尿嘧啶, 在DNA合成中发挥了束缚底物的关键功能。A20R赋予了全酶持续性合成的能力, 其能够募集DNA复制所需其他酶类[17]。
3.2.1 DNA聚合酶抑制剂DNA聚合酶在病毒DNA复制的过程中发挥着重要作用, 是抗病毒药物开发的有效靶点。该靶点抑制剂主要包括核苷类似物和无环磷酸核苷类似物(acyclic nucleoside phosphonates, ANPs), 其作用机制为在细胞内转化为磷酸盐的形式, 与脱氧胞苷三磷酸(dCTP)竞争, 在DNA聚合酶水平阻碍病毒DNA的合成。
3.2.1.1 核苷类似物核苷类似物进入细胞后需要经过痘病毒胸苷激酶(TK)催化的初始磷酸化步骤, 早期发现的代表性化合物有不同取代的胸腺嘧啶核苷(4, 图 3), 如化合物5~7 (图 3)等, 其活性如表 2所示, 活性明显优于CDV[18]。

|
Figure 3 Structures of thymidine analogues |
| Table 2 Activity of thymidine analogs against vaccinia virus and cowpox virus |
2013年, Duraffour等[19]发现了一种1-甲基取代的4-硫代胸腺嘧啶核苷化合物KAY-2-41 (8)具有抗HSV-1活性。后续研究显示, 该化合物在体外纳摩尔浓度下能有效抑制VACV (EC50 = 0.80 μmol·L-1)和CPXV (EC50 = 0.48 μmol·L-1)的复制, 与CDV相比活性提高(VACV, EC50 = 8.2 μmol·L-1; CPXV, EC50 = 13.9 μmol·L-1)。该化合物还对CDV的耐药正痘病毒株具有一定活性, 例如, 其抑制VACV-WR (A314V)的EC50为0.44 μmol·L-1, 与CDV (EC50 = 64.7 μmol·L-1)相比活性显著提高。机制研究发现其抑制活性可能干扰核苷激酶[例如胸苷激酶(TK)]的初始激活步骤。
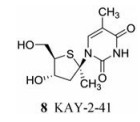
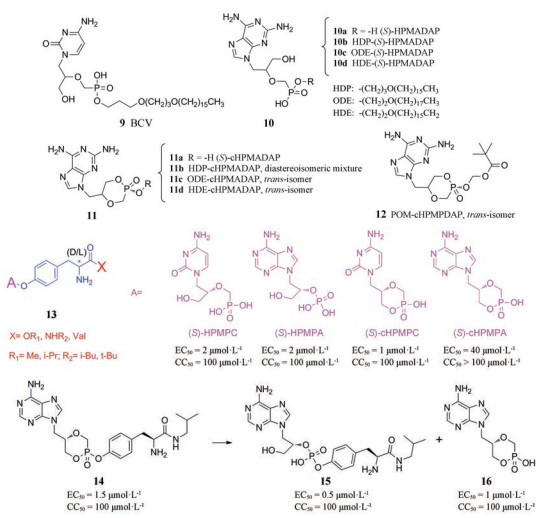
3.2.1.2 无环磷酸核苷类似物无环磷酸核苷类似物(ANPs)是一类重要的抗病毒药物, 其中3-羟基-2-(膦酰基甲氧基)丙基(HPMP)系列的无环核苷膦酸酯是最有希望的一类高活性、高选择性的抗痘病毒药物, 它包括各种碱基的衍生物, 如上市药物CDV及其衍生物、HPMPA及其衍生物、HPMPDAP及其衍生物以及HPMPO-DAPY等。该类化合物中磷酸基团的存在使其口服利用度普遍较差, 在其磷酸基团处引入烷基烷氧基侧链、形成环磷酸酯以及形成肽前药等都是改善口服利用度的有效策略。
|
Brincidofovir 在CDV的烷基烷氧基衍生物中, 前药brincidofovir (BCV, CMX001) (9), 是一种脂质共轭无环核苷酸磷酸酯。BCV一旦被细胞摄取, 其脂质部分裂解释放游离西多福韦, 进而抑制病毒DNA的合成[20]。早前研究证明BCV拥有对天花病毒(EC50 = 0.10 μmol·L-1)的体外活性, 与CDV (EC50 = 27.3 μmol·L-1)相比显著提高[21]。其口服利用度和肾毒性与CDV相比也显著改善[22]。2017年, Chittick等[23]经研究证明, 当以与治疗天花相似的剂量持续给药时, BCV在成人和儿童受试者中通常具有良好的耐受性, 最常见的不良反应是轻度胃肠道事件和血清转氨酶的无症状、短暂和可逆性升高。此外, 其安全性良好, 可以作为治疗天花的候选药物。
HPMPDAP酯类前药 2010年, Krecmerova等[24]通过对9-(S)-[3-羟基-2-(磷氧基)丙基]-2, 6-二氨基嘌呤(HPMPDAP)及其环状磷酸酯(cHPMPDAP)进行修饰, 合成了一系列烷氧基烷基、多肽类、新戊酰氧基甲酯(POM)、2, 2, 2-三氟乙基以及水杨酸衍生物, 其中HPMPDAP衍生物10b、10c和10d活性优于先导化合物10a。10b (EC50 = 0.000 74 μmol·L-1)、10c (EC50 = 0.000 74 μmol·L-1)、10d (EC50 = 0.001 2 μmol·L-1)相较于HPMPDAP (10a, EC50 = 0.50 μmol·L-1)活性增强了400~600倍。
cHPMPDAP衍生物11b (EC50 = 0.011 μmol·L-1)、11c (EC50 = 0.052 μmol·L-1)、11d (EC50 = 0.003 5 μmol·L-1)、12 (EC50 = 0.37 μmol·L-1)相较于先导化合物10a (EC50 = 0.53 μmol·L-1)活性增强了10倍[24]。因此HPMPDAP及其环型的烷氧基烷基和POM前药均具有良好的开发前景。
HPMPC和HPMPA氨基酸酯前药 为了克服烷氧基烷基衍生物毒性较大以及水溶性差等问题, 2011年, Zakharova等[25]合成了8种(S)-HPMPC和(S)-HPMPA及其环状磷酸酯[(S)-cHPMPC和(S)-cHPMPA]前药(13), 在磷酸基团的羟基处引入适当的氨基酸成酯, 以期达到改善口服利用度及增强抗病毒活性的目的。(S)-cHPMPC和(S)-cHPMPA的酪氨酸烷基酰胺酯具有更好的稳定性。研究发现酪氨酸酰胺衍生物活化途径简单, 其体外抗病毒活性与相应的母体药物相似。其中化合物14最稳定, 它在大鼠或小鼠血浆中仅转化为两种活性代谢物, 即化合物15和16, 在小鼠模型中口服生物利用度(39%)显著高于HPMPA (< 5%)。
|
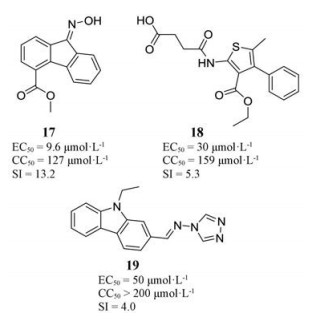
非核苷类抑制剂的发现扩大了痘病毒抑制剂的结构类型, 有利于克服现有核苷类药物的耐药性问题。2008年, Ciustea等[26]使用体外DNA合成快速平板测定法, 对49 663种化合物进行高通量筛选, 发现16种化合物具有体外抗痘苗病毒活性, 且细胞毒性较小。机制研究表明其抗病毒活性与干扰病毒DNA合成机制中的一个重要步骤相关。其中化合物17、18和19与CDV (EC50 = 132 μmol·L-1, CC50 > 500 μmol·L-1, SI > 3.8)相比, 活性明显增强且细胞毒性较小, 选择性指数(CC50/EC50)较高。
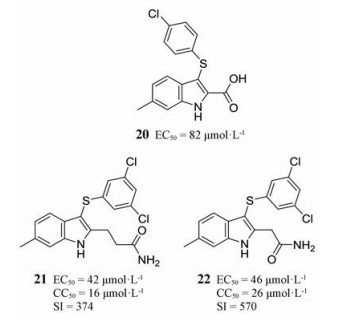
3.2.2 A20R-D4R二聚体抑制剂过程因子代表了一类独特的治疗靶点, 它们只与同源的DNA聚合酶相互作用, 具有高度的特异性。过程因子能够阻止DNA聚合酶从DNA模板链解离, 促进DNA链的持续性合成。痘病毒的过程因子是A20R和D4R两种蛋白构成的异二聚体[17], 目前研究较多的为化合物20及其衍生物, 它们可以在体外抑制DNA的合成。
|
2013年, Nuth等[27]通过先导化合物的吲哚2位的烷基胺修饰发现了化合物21和22, 其EC50分别为42和46 nmol·L-1, 显著优于先导化合物(EC50 = 82 μmol·L-1)。另外此课题组还建立了A20R-D4R的二聚体模型, 发现由于A20R-D4R蛋白的缔合作用, 其活性位点在两个蛋白交接处。化合物21和22通过烷基胺侧链向位点延伸从而破坏A20R-D4R的相互作用。化合物21和22作为一类新型的烷基胺取代吲哚衍生物, 具有广阔的研究前景。
|
在D4R、A20R和E9LDNA聚合酶构成的全酶中, D4R并不直接与E9L作用, A20R作为连接E9L和D4R的桥梁。D4R还具有尿嘧啶DNA糖基化酶(UDG)的活性, 这与其促进DNA合成的作用是相悖的。这也证明D4R作为过程因子在痘病毒DNA复制中扮演着重要的角色[17]。
2011年, Nuth等[28]在作用于A20R-D4R异二聚体的化合物库中通过高通量筛选筛选出27种化合物, 其EC50为9.6~23 μmol·L-1, 选择性指数为3.9~17.8, 抑制DNA合成和痘苗病毒感染的活性优于对照药物Fentichlor (DNA合成抑制剂) (EC50= 18.7 μmol·L-1)。通过热稳定性试验(ThermoFluor, 图 4A)发现化合物23~27 (图 4B)直接与D4R的UDG催化位点结合, 进而阻断DNA的合成, 最终有效抑制痘病毒感染。由于D4R的晶体结构已被确证, 可作为抗痘病毒药物研究的新靶点。

|
Figure 4 A: ThermoFluor, the hydrophobic dye, Sypro Orange, the unliganded (Tm1) and liganded complex (Tm2). B: The structures of compounds that target D4R |
痘病毒的转录可分为早期转录和晚期转录两个阶段, 其需要RNA聚合酶和众多转录因子的共同作用。其中A24R、A29L、A5R、D7R、E4L、G5R、H4L、J4R、J6R基因编码的蛋白以及加帽酶、甲基转移酶、ATP酶和DNA解旋酶等均可作为药物发现的靶点。尽管近年来已发现了一些转录抑制剂, 但大部分缺乏体内抗病毒活性。下文介绍了一些常见的痘病毒转录抑制剂。
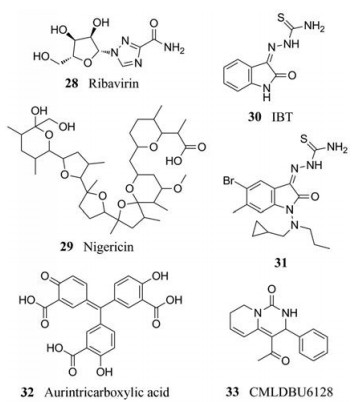
3.3.1 利巴韦林利巴韦林(ribavirin) (28)是一种广谱抗病毒药物, 对包括痘病毒在内的多种RNA和DNA病毒具有抑制活性, 但其在痘病毒的动物模型中没有效果。研究发现, 三磷酸利巴韦林是加帽酶的底物, 可以与mRNA5'末端共价连接, 影响翻译能力, 导致病毒基因表达效率低下[29]。这表明, mRNA分子的翻译可以被小分子抑制剂特异性阻断。
3.3.2 尼日利亚菌素尼日利亚菌素(29)是一种抗生素, 2010年, Myskiw等[30]发现, 尼日利亚菌素能够有效抑制痘苗病毒在几种人细胞系中的复制。在HeLa细胞中, 尼日利亚菌素的EC50为7.9 nmol·L-1, 与对照药物CDV (EC50 = 110 nmol·L-1)相比活性大大增强。研究发现其作用机制与痘病毒的早期转录和晚期转录过程相关。
|
早前发现的化合物2, 3-吲哚二酮3-硫代缩氨基脲(30)对痘苗病毒在体内和体外的复制均表现出良好的抑制活性, 但缺乏对牛痘病毒的体内抑制活性[31]。有研究发现, 其作用机制与转录过程相关[32]。2005年, Pirrung等[33]通过高通量筛选, 发现了30的衍生物31, 对VACV (EC50 = 0.6 μmol·L-1)和CPXV (EC50 = 6 μmol·L-1)具有很好的抑制活性, 与先导化合物IBT (VACV, EC50 = 14 μmol·L-1; CPXV, EC50 = 60 μmol·L-1)和阳性药物CDV (VACV, EC50 = 27 μmol·L-1; CPXV, EC50 = 38 μmol·L-1)相比活性显著提高。
3.3.4 其他化合物一些抑制转录但缺乏体内有效性的化合物也已被发现, 如金精三羧酸(31)能抑制正痘病毒的早期转录[34]。另外, 一种非核苷抑制剂CMLDBU6128 (32)也可以抑制正痘病毒晚期转录中基因的表达[35]。
3.4 病毒粒子的组装和释放抑制剂在细胞内, D13蛋白与一组病毒蛋白形成病毒膜组装蛋白, 然后与从内质网衍生出的膜前体, 组装成新月体, 新月体包裹病毒核心产生未成熟病毒粒子(Ⅳ)[36]。未成熟病毒粒子经历病毒核心的缩合, 蛋白水解, 支架的丧失和形成以及形成新的二硫键等过程, 成为成熟的病毒粒子即IMV结构[37]。
IMV通过复杂的包裹机制获得第二个包膜, 并最终作为EEV释放。该过程依赖于病毒膜蛋白A27L、E2L、F13L和B5R, 这些蛋白只存在于EFV中, 是EEV的重要生物学特征[38]。IEV通过微管主动转运至细胞膜, 并与细胞膜融合, 通过胞吐作用在细胞表面形成CEV。CEV离开细胞, 形成EEV[39]。
3.4.1 D13抑制剂病毒组装所需要的膜前体是开放式管状结构, 其由于病毒蛋白A17的网状样作用而产生明显的曲率。A17能够将D13募集到病毒包膜上。早期研究发现抗生素利福平(34, 图 5A)抑制正痘病毒的复制, 其靶向于D13, 抑制新月体的形成[40]。

|
Figure 5 A: The structure of rifampin and the docking diagram of rifampin and D13. B: Stereoview of the rifampicin binding site. C: The detailed binding mode of rifampin with D13. The naphtoquinone core is colored in blue; ansa bridge is in pink, and C3 piperazine branch is in green. An asterisk indicates the C3 carbon |
2018年, Garriga等[41]通过测定D13与利福平复合物的晶体结构(PDB code:6beb), 发现利福平在D13三聚体中央通道的近膜开口处与富含苯丙氨酸的区域(F环)结合(图 5B)。如图 5C所示, 萘醌核的平面表面占据了通道的大部分, C3支链向外。连接萘醌核心两边的长链, 称为ansa桥, 朝向通道内侧, 它与E165/A残基之间水桥介导的氢键, 形成了利福平和D13之间仅有的极性相互作用。利用X射线晶体学、表面等离子共振(SPR)、NMR和定点诱变, 证明D13上A17的结合位点与利福平的结合位点重叠, 表明利福平干扰D13与A17的相互作用, 从而阻止未成熟病毒粒子的形成。
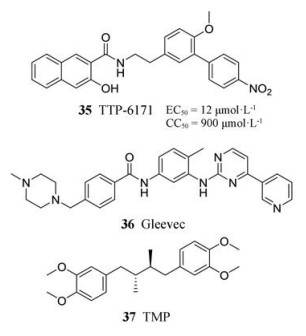
3.4.2 I7L抑制剂痘病毒I7L泛素样蛋白酶(ULP)对病毒复制至关重要, 在痘病毒科中高度保守, 是开发抗天花病毒药物的一个潜力靶点。目前已发现一种特异性的I7L抑制剂TTP-6171 (EC50 = 12 μmol·L-1, CC50 = 900 μmol·L-1) (35), 与CDV (EC50 = 33 μmol·L-1, CC50 = 280 μmol·L-1)相比活性增强, 且细胞毒性显著降低[42], 是一类非常有潜力的抗正痘病毒药物先导化合物。
3.4.3 F13L抑制剂F13L包膜蛋白是病毒粒子释放过程中的一个重要靶点, 痘病毒F13L编码细胞外病毒颗粒形成所需的37 kDa的棕榈酰化外周膜蛋白。该蛋白质参与细胞内成熟病毒(IMV)颗粒的包裹, 以产生感染性病毒颗粒[10]。如前所述, 上市药物ST-246靶向于F13L。
3.5 病毒传播抑制剂IEV粒子与宿主细胞的质膜融合, 形成细胞相关的包膜病毒(CEV)。CEV可以直接脱离细胞, 也可以利用Abl和Src家族酪氨酸激酶诱导肌动蛋白聚合反应, 脱离细胞, 形成细胞外被膜病毒(EEV)[43]。IMV在体内容易被宿主免疫系统发现和灭活, 而CEV和EEV逃避免疫检测, 介导了感染的传播。
3.5.1 伊马替尼伊马替尼(36)是一种用于治疗人类慢性粒细胞白血病的Abl-家族的激酶抑制剂。研究表明, 伊马替尼在感染痘病毒的小鼠中, 能够有效抑制病毒的传播[43]。
3.5.2 TerameprocolTerameprocol (TMP, 37)是去甲二氢愈创木酚酸(nordihydroguaiaretic acid)的甲基化衍生物。2010年, Pollara等[44]发现TMP在多种细胞系中均能有效抑制牛痘病毒的生长, 且能使感染细胞表面的痘病毒肌动蛋白尾部明显减少, 这表明其可能通过抑制肌动蛋白的形成, 从而阻止病毒在细胞间的传播。实验结果表明, TMP在所有测试的细胞系中均抑制了牛痘病毒的复制, 对每一种细胞类型, 测定感染后5、12、24和48 h的病毒产量, 计算48 h后经TMP处理的细胞与经DMSO处理的细胞相比, 牛痘病毒生长的抑制率达到30%~99.9%。这也表明痘病毒发病机制与不同细胞间的传播机制密切相关。
|
N1L蛋白是一种牛痘病毒和天花病毒毒力因子, 具有Bcl-2家族蛋白的BH3肽结合域特征, 能够与BH3肽结合。NIL蛋白并不是病毒复制的关键因子, 但是当其缺失时, 培养物中病毒产量会大幅度衰减。并且有研究表明, 它参与调节细胞凋亡机制[45]。
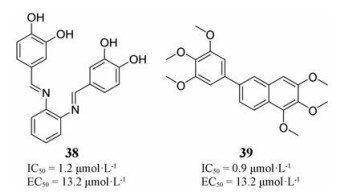
2010年, Cheltsov等[46]通过测定化合物与NIL蛋白的亲和力, 发现了亚微摩尔级别的N1L拮抗剂, 如化合物38和39, 其亲和力可与BH3肽(IC50 = 70~1 000 nmol·L-1)相媲美, 在体外能够抑制牛痘病毒的复制。因此对N1L凋亡阻断的特异性进行抑制可以作为抗痘病毒感染的一种治疗策略。
|
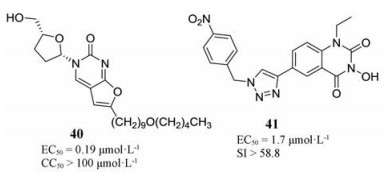
2013年, Mcguigan等[47]通过对L-双脱氧双环核苷类似物(L-ddBCNAs)的活性构效关系研究, 发现具有醚类侧链的化合物对痘苗病毒(VACV)的体外抑制活性显著增强, 化合物40 (EC50 = 0.19 μmol·L-1)活性最优, 与CDV (EC50 = 11.5 μmol·L-1)相比提高了60倍, 但其作用机制尚不清楚。
4.3 喹诺酮类化合物2015年, 本课题组利用点击化学合成了一系列新型的1, 2, 3-三唑基-3-羟基-喹唑啉-2, 4(1H, 3H)-二酮类化合物, 在体外对痘病毒和腺病毒具有较好的抑制活性, 其中化合物41对牛痘病毒的抑制作用最强, EC50值为1.7 μmol·L-1, 是对照药物CDV (EC50 = 25 μmol·L-1, SI > 10)的15倍[48], 有望作为治疗痘病毒感染的候选药物进行研究。
5 总结与展望痘病毒作为已知的病毒粒子中结构最复杂、宿主范围最广和成员最多的病毒家族, 其多数成员都能引起动物源性的人畜共患病, 而天花暴发也曾对人类造成严重的伤害。由于全球天花的消灭, 抗天花的疫苗接种间断, 致使全球对天花易感的人群逐渐增加。当前, 全球范围都面临着包括天花在内的生物武器袭击的威胁。因此, 积极寻找能够有效的抗痘病毒新药依然具有重要意义。ST-246的获批对痘病毒潜在的生物威胁起到有效预防, 其与CDV不同的作用机制, 也为新型抑制剂的研发提供了可能。
|
本文总结了病毒复制周期各阶段中已有研究的靶点及其抑制剂, 这些靶点主要是各种酶类和相关的基因产物。近年来, 抗痘病毒药物的研究着重于上市药物和候选药物的修饰、潜力靶点抑制剂的研究以及新的靶点的开发。通过前药策略、老药新用、化合物库的构建与表型筛选发现了许多结构新颖的痘病毒抑制剂, 并确定了部分化合物的作用靶点。
核苷类似物一直是抗病毒药物研究的热点, HPMP系列的无环核苷磷酸酯(ANPs)是最有希望的一种高效、选择性的抗痘病毒药物, 近年来利用前药策略降低核苷类化合物的毒性并提高其口服利用度, 涌现出了大量有效的正痘病毒抑制剂。根据美国FDA的动物法规, 目前正在开发brincidofovir作为治疗天花的候选药物, 其优越的安全性、广谱性、良好的口服生物利用度和在动物模型中的有效性, 使其很有可能被批准用于治疗痘病毒感染。随着分子生物学、细胞生物学、结构生物学等学科的快速发展, 相信会有更多高特异性的新型抗痘病毒药物上市。
| [1] |
Essbauer S, Pfeffer M, Meyer H. Zoonotic poxviruses[J]. Vet Microbiol, 2010, 140: 229-236. DOI:10.1016/j.vetmic.2009.08.026 |
| [2] |
Voigt EA, Kennedy RB, Poland GA. Defending against smallpox: a focus on vaccines[J]. Expert Rev Vaccines, 2016, 15: 1197-1211. DOI:10.1080/14760584.2016.1175305 |
| [3] |
Kupferschmidt K. Labmade smallpox is possible, study shows[J]. Science, 2017, 357: 115-116. DOI:10.1126/science.357.6347.115 |
| [4] |
Jing ZZ, Jia HJ, Zhou T. Poxvirus disease: a kind of zoonoses worthy of high attention[J]. Chin J Viral Dis (中国病毒病杂志), 2011, 1: 416-418. |
| [5] |
Gunther T, Haas L, Alawi M, et al. Recovery of the first full-length genome sequence of a parapoxvirus directly from a clinical sample[J]. Sci Rep, 2017, 7: 3734. |
| [6] |
Roberts KL, Smith GL. Vaccinia virus morphogenesis and dissemination[J]. Trends Microbiol, 2008, 16: 472-479. DOI:10.1016/j.tim.2008.07.009 |
| [7] |
Julien O, Beadle J R, Magee W C, et al. Solution structure of a DNA duplex containing the potent anti-poxvirus agent cidofovir[J]. J Am Chem Soc, 2011, 133: 2264-2274. DOI:10.1021/ja109823e |
| [8] |
De Clercq E, Holy A. Acyclic nucleoside phosphonates: a key class of antiviral drugs[J]. Nat Rev Drug Discov, 2005, 4: 928-940. |
| [9] |
Hoy SM. Tecovirimat: first global approval[J]. Drugs, 2018, 78: 1377-1382. DOI:10.1007/s40265-018-0967-6 |
| [10] |
Duraffour S, Lorenzo MM, Zoller G, et al. ST-246 is a key antiviral to inhibit the viral F13L phospholipase, one of the essential proteins for orthopoxvirus wrapping[J]. J Antimicrob Chemother, 2015, 70: 1367-1380. DOI:10.1093/jac/dku545 |
| [11] |
Carter GC, Law M, Hollinshead M, et al. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans[J]. J Gen Virol, 2005, 86: 1279-1290. DOI:10.1099/vir.0.80831-0 |
| [12] |
Brown E, Senkevich TG, Moss B. Vaccinia virus F9 virion membrane protein is required for entry but not virus assembly, in contrast to the related L1 protein[J]. J Virol, 2006, 80: 9455-9464. DOI:10.1128/JVI.01149-06 |
| [13] |
Ojeda S, Senkevich TG, Moss B. Entry of vaccinia virus and cell-cell fusion require a highly conserved cysteine-rich membrane protein encoded by the A16L gene[J]. J Virol, 2006, 80: 51-61. |
| [14] |
Senkevich TG, Ojeda S, Townsley A, et al. Poxvirus multiprotein entry-fusion complex[J]. Proc Natl Acad Sci U S A, 2005, 102: 18572-18577. DOI:10.1073/pnas.0509239102 |
| [15] |
Bell E, Shamim M, Whitbeck JC, et al. Antibodies against the extracellular enveloped virus B5R protein are mainly responsible for the EEV neutralizing capacity of vaccinia immune globulin[J]. Virology, 2004, 325: 425-431. DOI:10.1016/j.virol.2004.05.004 |
| [16] |
Demasi J, Du S, Lennon D, et al. Vaccinia virus telomeres: interaction with the viral I1, I6, and K4 proteins[J]. J Virol, 2001, 75: 10090-10105. |
| [17] |
Boyle KA, Stanitsa ES, Greseth MD, et al. Evaluation of the role of the vaccinia virus uracil DNA glycosylase and A20 proteins as intrinsic components of the DNA polymerase holoenzyme[J]. J Biol Chem, 2011, 286: 24702-24713. |
| [18] |
Prichard MN, Kern ER. Antiviral activity of 4'-thioIDU and thymidine analogs against orthopoxviruses[J]. Viruses, 2010, 2: 1968-1983. |
| [19] |
Duraffour S, Drillien R, Andrei G, et al. KAY-2-41, a novel nucleoside analogue inhibitor of orthopoxviruses in vitro and in vivo[J]. Antimicrob Agents Chemother, 2014, 58: 27-37. |
| [20] |
Magee WC, Hostetler KY, Evans DH. Mechanism of inhibition of vaccinia virus DNA polymerase by cidofovir diphosphate[J]. Antimicrob Agents Chemother, 2005, 49: 3153-3162. DOI:10.1128/AAC.49.8.3153-3162.2005 |
| [21] |
Lanier R, Trost L, Tippin T, et al. Development of CMX001 for the treatment of poxvirus infections[J]. Viruses, 2010, 2: 2740-2762. |
| [22] |
Tippin TK, Morrison ME, Brundage TM, et al. Brincidofovir is not a substrate for the human organic anion transporter 1: a mechanistic explanation for the lack of nephrotoxicity observed in clinical studies[J]. Ther Drug Monit, 2016, 38: 777-786. |
| [23] |
Chittick G, Morrison M, Brundage T, et al. Short-term clinical safety profile of brincidofovir: a favorable benefit-risk proposition in the treatment of smallpox[J]. Antiviral Res, 2017, 143: 269-277. |
| [24] |
Krecmerova M, Holy A, Andrei G, et al. Synthesis of ester prodrugs of 9-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]-2, 6-diaminopurine (HPMPDAP) as anti-poxvirus agents[J]. J Med Chem, 2010, 53: 6825-6837. |
| [25] |
Zakharova VM, Serpi M, Krylov IS, et al. Tyrosine-based 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]cytosine and -adenine ((S)-HPMPC and (S)-HPMPA) prodrugs: synthesis, stability, antiviral activity, and in vivo transport studies[J]. J Med Chem, 2011, 54: 5680-5693. |
| [26] |
Ciustea M, Silverman JE, Druck Shudofsky AM, et al. Identification of non-nucleoside DNA synthesis inhibitors of vaccinia virus by high-throughput screening[J]. J Med Chem, 2008, 51: 6563-6570. DOI:10.1021/jm800366g |
| [27] |
Nuth M, Guan H, Zhukovskaya N, et al. Design of potent poxvirus inhibitors of the heterodimeric processivity factor required for viral replication[J]. J Med Chem, 2013, 56: 3235-3246. |
| [28] |
Nuth M, Huang L, Saw YL, et al. Identification of inhibitors that block vaccinia virus infection by targeting the DNA synthesis processivity factor D4[J]. J Med Chem, 2011, 54: 3260-3267. |
| [29] |
Bougie I, Bisaillon M. The broad spectrum antiviral nucleoside ribavirin as a substrate for a viral RNA capping enzyme[J]. J Biol Chem, 2004, 279: 22124-22130. |
| [30] |
Myskiw C, Piper J, Huzarewich R, et al. Nigericin is a potent inhibitor of the early stage of vaccinia virus replication[J]. Antiviral Res, 2010, 88: 304-310. DOI:10.1016/j.antiviral.2010.10.001 |
| [31] |
Quenelle DC, Keith KA, Kern ER. In vitro and in vivo evaluation of isatin-beta-thiosemicarbazone and marboran against vaccinia and cowpox virus infections[J]. Antiviral Res, 2006, 71: 24-30. |
| [32] |
Cresawn SG, Condit RC. A targeted approach to identification of vaccinia virus postreplicative transcription elongation factors: genetic evidence for a role of the H5R gene in vaccinia transcription[J]. Virology, 2007, 363: 333-341. |
| [33] |
Pirrung MC, Pansare SV, Sarma KD, et al. Combinatorial optimization of isatin-beta-thiosemicarbazones as anti-poxvirus agents[J]. J Med Chem, 2005, 48: 3045-3050. DOI:10.1021/jm049147h |
| [34] |
Myskiw C, Deschambault Y, Jefferies K, et al. Aurintricarboxylic acid inhibits the early stage of vaccinia virus replication by targeting both cellular and viral factors[J]. J Virol, 2007, 81: 3027-3032. |
| [35] |
Dower K, Filone CM, Hodges EN, et al. Identification of a pyridopyrimidinone inhibitor of orthopoxviruses from a diversity-oriented synthesis library[J]. J Virol, 2012, 86: 2632-2640. DOI:10.1128/JVI.05416-11 |
| [36] |
Maruri-Avidal L, Weisberg AS, Moss B. Direct formation of vaccinia virus membranes from the endoplasmic reticulum in the absence of the newly characterized L2-interacting protein A30.5[J]. J Virol, 2013, 87: 12313-12326. |
| [37] |
Byrd CM, Hruby DE. Vaccinia virus proteolysis--a review[J]. Rev Med Virol, 2006, 16: 187-202. DOI:10.1002/rmv.499 |
| [38] |
Perdiguero B, Lorenzo MM, Blasco R. Vaccinia virus A34 glycoprotein determines the protein composition of the extracellular virus envelope[J]. J Virol, 2008, 82: 2150-2160. DOI:10.1128/JVI.01969-07 |
| [39] |
Newsome TP, Scaplehorn N, Way M. SRC mediates a switch from microtubule- to actin-based motility of vaccinia virus[J]. Science, 2004, 306: 124-129. DOI:10.1126/science.1101509 |
| [40] |
Charity JC, Katz E, Moss B. Amino acid substitutions at multiple sites within the vaccinia virus D13 scaffold protein confer resistance to rifampicin[J]. Virology, 2007, 359: 227-232. |
| [41] |
Garriga D, Headey S, Accurso C, et al. Structural basis for the inhibition of poxvirus assembly by the antibiotic rifampicin[J]. Proc Natl Acad Sci U S A, 2018, 115: 8424-8429. DOI:10.1073/pnas.1810398115 |
| [42] |
Byrd CM, Bolken TC, Mjalli AM, et al. New class of orthopoxvirus antiviral drugs that block viral maturation[J]. J Virol, 2004, 78: 12147-12156. DOI:10.1128/JVI.78.22.12147-12156.2004 |
| [43] |
Reeves PM, Bommarius B, Lebeis S, et al. Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases[J]. Nat Med, 2005, 11: 731-739. DOI:10.1038/nm1265 |
| [44] |
Pollara JJ, Laster SM, Petty IT. Inhibition of poxvirus growth by terameprocol, a methylated derivative of nordihydroguaiaretic acid[J]. Antiviral Res, 2010, 88: 287-295. DOI:10.1016/j.antiviral.2010.09.017 |
| [45] |
Bartlett N, Symons JA, Tscharke DC, et al. The vaccinia virus N1L protein is an intracellular homodimer that promotes virulence[J]. J Gen Virol, 2002, 83: 1965-1976. DOI:10.1099/0022-1317-83-8-1965 |
| [46] |
Cheltsov AV, Aoyagi M, Aleshin A, et al. Vaccinia virus virulence factor N1L is a novel promising target for antiviral therapeutic intervention[J]. J Med Chem, 2010, 53: 3899-3906. |
| [47] |
Mcguigan C, Hinsinger K, Farleigh L, et al. Novel antiviral activity of L-dideoxy bicyclic nucleoside analogues versus vaccinia and measles viruses in vitro[J]. J Med Chem, 2013, 56: 1311-1322. |
| [48] |
Kang D, Zhang H, Zhou Z, et al. First discovery of novel 3-hydroxy-quinazoline-2, 4(1H, 3H)-diones as specific anti-vaccinia and adenovirus agents via 'privileged scaffold' refining approach[J]. Bioorg Med Chem Lett, 2016, 26: 5182-5186. |