 2020, Vol. 55
2020, Vol. 55



腺病毒(adenovirus, Ad)是腺病毒科成员之一, 于1953年首次从腺样体切除术中获得的人腺样体中分离出来, 根据其生物学、理化和遗传特性不同, 目前已发现超过100种Ad血清型, 这些血清型被进一步划分为从A到G共7个亚群, 其中人腺病毒有52种, 分别命名为Adl~Ad52, 分为A、B、C、D、E和F六个亚群。
腺病毒是一种无包膜的裂解病毒, 具有独特的二十面体形态(图 1)[1]。成熟的病毒粒子直径约为70~90 nm, 由蛋白衣壳、病毒DNA及核心蛋白组成。其中, 病毒蛋白衣壳由六邻体、五邻体和纤突3种主要的蛋白构成, 共252个壳粒, 二十面体的12个顶角壳粒为五邻体, 五邻体上有纤突, 剩下240个非顶角壳粒为六邻体。六邻体是构成衣壳最丰富的衣壳蛋白, 它是病毒多肽蛋白PⅡ的同源三聚体, 由三角形的塔尖和五面体的基底组成, 塔区由4个环构成即loop 1~4, 基底包含两个区域P1、P2区, loop 1上有6个高变区, loop 2上有1个高变区, 7个高变区含有血清型特异的抗原决定簇, 是诊断不同血清型的标准; 多肽PVI、PⅦI在六邻体与病毒核心之间形成连接桥, 并与多肽PIX一起维持着六邻体分子的晶格排列的稳定。每个五邻体基底由5个多肽PIII相连构成, 上面结合着1根(哺乳动物腺病毒)或2根(禽腺病毒)长9~77. 5 nm的纤维突起, 这些纤突以五邻体蛋白为基底由衣壳面伸出, 顶端形成头节区, 主要由多肽蛋白PIV组成, 纤突有血清特异性, 且含有负责体外血细胞凝集的种属特异性抗原决定位点; 多肽PIIIa为五邻体的周围蛋白, 也参与衣壳的组成, 五邻体通过多肽PV与病毒核心相连。

|
Figure 1 Schematic view of adenovirus structure |
每个病毒颗粒包含一个36 kb的线性双链DNA, 两端各有一个100~600 bp的反向末端重复序列(inverted terminal repeat, ITR), 是复制的起始位点, ITR的内侧为病毒包装信号(ψ), 介导腺病毒基因包装入病毒衣壳, 两端的ITR和ψ是病毒包装所需要的顺式作用元件。基因组包含早期表达的与腺病毒复制相关的E1~E4基因和晚期表达的与腺病毒颗粒组装相关的L1~L5基因, 线状双股DNA被蛋白Ⅶ和mu蛋白紧密地环绕, 形成直径为60~65 nm的髓芯, 被包裹于衣壳内。另一种蛋白V将这种DNA-蛋白复合物连接起来, 并通过蛋白Ⅵ与病毒衣壳连接在一起。双链DNA的5′端与DNA末端前体蛋白(precursor terminal protein, pTP)以磷酸二酯键结合生成复合物。
腺病毒的生命周期主要分为吸附、侵入、转录、早期表达、复制、晚期表达、组装与释放等过程(图 2)[2]。腺病毒感染细胞的过程是从腺病毒纤毛的头节区吸附到细胞表面的特异性受体开始的, 接下来病毒纤毛基底部五邻体表面的三肽RGD与细胞表面的αvβ3和αvβ5整合素结合, 通过内吞作用将腺病毒内化到细胞中并进入溶酶体; 在溶酶体的酸性环境下, 腺病毒衣壳的构象发生变化, 释放蛋白Ⅵ的顶体蛋白, 蛋白Ⅵ能够破坏胞内体膜, 允许部分分解的病毒颗粒从胞内体中逃逸; 去包装的病毒通过动力蛋白沿着微管被运送到复杂的核心孔, 病毒DNA通过复杂的核心孔被输送入核心。病毒基因组进入细胞核后, 细胞转录因子首先与E1A区上游的增强子结合, 表达E1A蛋白, 该蛋白的作用是调节细胞代谢, 使病毒DNA更易在细胞中复制, E1A蛋白还可以激活其他早期基因(E1B、E2A、E2B、E3和E4)的启动子, 其中E2B驱动另外3个与病毒复制有关的早期基因转录单位末端前体蛋白pTP、单链DNA结合蛋白(single-stranded DNA binding proteins, ssDBP)以及DNA聚合酶(DNA polymerase, DNA pol)的表达, 这3个基因的表达产物紧密地结合成一个复合物, 反活化其他蛋白, 启动病毒基因组的复制。感染约7 h后, 腺病毒的DNA复制由蛋白启动, 首先在ITRs中pTP与DNA聚合酶相互作用形成一个紧密结合的起始前复合物, 之后一段与模板链4~6位核苷酸相反的CAT三核苷酸共价偶联到pTP的丝氨酸残基上, pTP-CAT中间体介导模板的前3个核苷酸的碱基配对, 而进一步的延伸需要DNA结合蛋白和DNA聚合酶进行链置换合成, 置换出的单链分子可以自我退火环化, 形成锅柄样环形分子, 然后这种环形分子再以相同的机制合成出子代双链DNA分子。复制的同时, 早期基因的转录和翻译被关闭, 晚期基因开始表达。大部分晚期基因的转录是由一个共同的主要晚期启动子(major late promoter, MLP)调控的。实际上, MLP的活性与病毒基因组复制密切相关, 一旦腺病毒基因组开始复制, MLP的活性将明显增强, 晚期基因主要编码腺病毒的结构蛋白及包膜。之后病毒结构蛋白在细胞核内聚集形成病毒衣壳, 病毒的基因组被包装进去, 形成有感染能力的病毒颗粒, 并最终通过裂解宿主细胞被释放出去, 完成腺病毒的生命周期[3]。

|
Figure 2 Schematic view of the life cycle of adenovirus |
腺病毒受体包括柯萨奇-腺病毒受体(Coxsackic-adenovirus receptor, CAR)、CD46、CD80、CD86以及唾液酸受体。除B和D亚种外, 其他亚种的受体都是柯萨奇-腺病毒受体, 它是柯萨奇病毒与腺病毒的特异性受体, 其本质是一种细胞间黏附分子, 在上皮细胞的紧密连接和细胞间相互连接处表达, 贯通细胞膜, 分为胞外区、跨膜区、胞内区; CAR的表达水平在介导腺病毒感染方面起着重要作用, 其表达在体内是可调节的, 具有阶段依赖性, CAR受体表达越高, 该组织器官越容易被感染; 大多数B亚种可与胞膜表面免疫调节分子CD46相结合, 但Ad3和Ad7分别与相关免疫调节分子CD80或CD86结合, D亚种可与细胞表面普遍存在的唾液酸受体结合[4]。另一种参与腺病毒感染细胞的介质是细胞表面的第二受体整合素, 包括ανβ3、ανβ5、ανβ1、α3β1和α5β1, 其中最具特色的是ανβ3与ανβ5, 除HAdV-F40、HAdV-F41以外, 其他腺病毒血清型的五邻体基座上都有整合素的识别位点—RGD肽, 整合素与五邻体基座里包含有RGD肽的外环相互作用有助于腺病毒进入宿主细胞, 整合素在细胞表面表达水平的高低, 会影响腺病毒的感染效率。由于Ad血清型对应的受体及受体的体内分布不同, 导致腺病毒血清型间的组织趋向性不同, C、E和一些B亚种血清型腺病毒通常导致呼吸道感染, 其他B亚种血清型会导致泌尿道感染, A和F亚种血清型主要导致胃肠道感染, D亚种血清型则导致眼部感染[5]。
腺病毒是一种常见的机会性病原体, 在健康人中很少引起严重的临床症状, 但对于艾滋病患者、遗传性免疫缺陷患者以及接受免疫抑制治疗的实体器官或造血干细胞移植患者等免疫功能低下的人群, 可引起严重症状, 甚至致命。腺病毒可导致的临床疾病包括呼吸系统感染、胃肠炎、肝炎、脑炎、结膜炎、出血性膀胱炎、肾炎和免疫缺陷患者(包括艾滋病毒感染者)的感染性腹泻。在过去的20年中, 由于器官移植手术的增多, 伴随着强有力的免疫抑制疗法, 以及艾滋病毒的流行, 严重和危及生命的腺病毒感染的发生率逐渐增加, 但抗腺病毒感染的治疗仍不令人满意[6]。
目前临床上还没有针对腺病毒感染的特异性药物, 一般采用广谱抗病毒治疗和免疫治疗。抗病毒治疗主要局限于如更昔洛韦、阿昔洛韦、阿糖腺苷、利巴韦林和西多福韦等非特异性用药, 目前西多福韦、利巴韦林和更昔洛韦等广谱抗病毒药物已经在体外实验和临床环境中进行了抗腺病毒活性测试, 但效果差异大, 没有显示出令人满意的结果。因此迫切需要发现特异性、安全性和生物利用度高的抗腺病毒药物和疫苗[1]。
抗腺病毒药物的机制主要包括以下几种: ①药物与腺病毒表面特定部位结合, 在病毒吸附细胞的过程中, 药物可能会破坏腺病毒的纤突, 阻止病毒的吸附, 使病毒不能进入细胞内; ②药物与细胞表面受体结合, 改变细胞构象, 使对腺病毒敏感的细胞变为非敏感细胞, 从而改变细胞膜上病毒的数量和结构来控制病毒的感染; ③药物的某些成分进入细胞内, 对胞内基因的代谢和表达起调节作用, 从而抑制病毒核酸复制或阻止病毒蛋白合成[6]。
本文根据腺病毒的复制周期及治疗靶点, 精选近几年研究实例, 从药物化学的角度总结了抗腺病毒药物新靶标及其抑制剂的前沿进展。
1 病毒吸附抑制剂腺病毒吸附过程依赖病毒表面的纤维蛋白与细胞受体之间的相互识别和相互作用, 是一个有吸引力的靶点。腺病毒体的纤维旋钮作为受体结合域, 位于每个纤维的C端, 包含3个独立的口袋, 每个口袋可以容纳1个唾液酸残基。Ad37等亚型的受体被证明是细胞表面的一种糖蛋白, 这些糖蛋白携带一种类似GD1a神经节苷元的糖基结构, GD1a聚糖是一种分支的六糖, 其中两个分支上末端有唾液酸残基, 这两个唾液酸分子与Ad37纤维蛋白的三聚体中的3个唾液酸结合位点中的两个相连接, 所以在吸附过程中, 唾液酸起主要调节作用。从Ad37纤维蛋白中与唾液酸相互作用的纤维旋钮结构域与唾液酸的共晶结构看, 唾液酸与蛋白之间存在的相互作用主要包括盐桥、氢键(两种)和疏水作用。2位直立键上的羧酸与Lys345的氨基形成盐桥, 4位羟基与Tyr312的羟基形成氢键, 酰胺基通过氢键与Pro317的羰基结合, 然后唾液酸的乙酰基直接进入由Tyr312、Tyr308与另一种蛋白质单体Pro317和Val322形成的深疏水性口袋中(图 3)[7]。因此, 用天然或合成的唾液酸衍生物抑制腺病毒可能会阻止病毒粒子附着、渗透及感染新细胞, 并且利用Ad37纤维旋钮处的三聚体结合位点可克服单价唾液酸衍生物的相对低效的缺点, 与唾液酸单糖相比, 多价态唾液酸衍生物或糖缀合物可以同时结合到每个纤维旋钮的几个唾液酸识别口袋, 可大大提高抑制活性。

|
Figure 3 View of the sialic acid-interacting knob domain of the Ad37 fiber protein in complex with sialic acid. Four main interactions between the Ad37 fiber protein and the sialic acid are highlighted: two hydrogen bonds (4-OH-Tyr312, 5-NH-Pro317), one salt bridge (COO-Lys345), and one hydrophobic interaction between the N-acetyl group of sialic acid and the hydrophobic pocket generated by Tyr312 and Pro317 (PDB code: 1UXA). The pictures were made by Pymol |
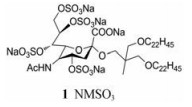
Kaneko等[8]报道硫酸唾液酸衍生物1 (NMSO3)作为病毒吸附抑制剂可以与唾液酸化细胞受体竞争结合腺病毒颗粒, 阻止腺病毒吸附到易感细胞上, 被发现在不影响细胞存活的复合浓度下抑制几种腺病毒血清型的细胞结合, 其有效性和选择性均优于西多福韦(HPMPC)与扎西他滨(ddC)。
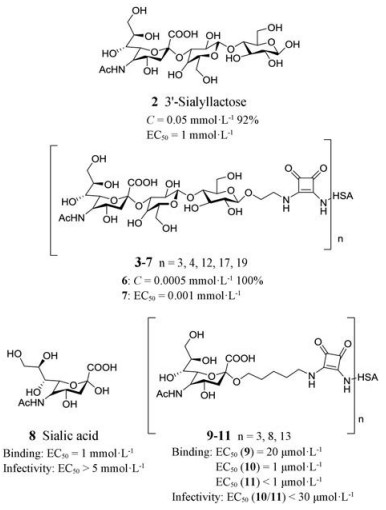
1.2 多价HSA靶向唾液酸偶联物2005年, Johansson等[9]设计合成了3'-唾液酸乳糖的多价HSA偶联物3~7, 并报道它们可作为腺病毒吸附和感染的有效抑制剂, 有效地阻止Ad37与人角膜上皮(HCE)细胞的结合和感染, 并且随着价态水平的增加, 抑制作用明显增强。其中在Ad37对HCE细胞的结合抑制试验中, 化合物6 (17价3'-唾液酸乳糖HSA偶联物)浓度为0.000 5 mmol·L-1时, 抑制率可达100%, 抑制活性是2 (单价唾液酸乳糖偶联物) (浓度为0.05 mmol·L-1时, 抑制率达92%)的100多倍; 在Ad37对HCE细胞的感染抑制试验中, 化合物7 (19价3'-唾液酸乳糖HSA偶联物)的抑制活性是2的1 000倍左右。随后, Johansson等[10]继续探讨了多价唾液酸偶联物对Ad37与人角膜上皮细胞结合及感染的抑制作用, 设计合成了唾液酸的多价HSA偶联物9~11, 其中在Ad37对HCE细胞的结合抑制试验中, 10 (8价唾液酸HSA偶联物)和11 (13价唾液酸HSA偶联物)的活性约是8 (单价唾液酸)的100倍; 在Ad37对HCE细胞的感染抑制试验中, 10和11的活性同样较8提高了100多倍。这些结果证实了价态水平决定多价唾液酸衍生物对腺病毒吸附和感染的抑制活性水平。多价唾液酸偶联物较多价唾液酸乳糖偶联物相比, 具有结构体积小, 复杂性低, 合成容易, 更利于进一步结构优化以及成本更低的优势。
|
|
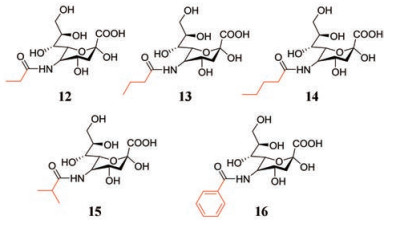
从纤维旋钮蛋白与唾液酸内酯的晶体结构来看, 唾液酸乙酰胺基明显位于一个较大的疏水囊中, 为了提高多价唾液酸偶联物的效力, Johansson等[7]采用了基于结构的药物化学设计策略, 保持唾液酸的骨架结构不变, 通过优化疏水口袋与5′-酰胺之间的相互作用, 合成了一系列N-酰基修饰的唾液酸衍生物, 以提高化合物与口袋的疏水相互作用, 从而提高亲和力和功效。通过基于蛋白-配体相互作用的计算机模拟分析可知只有具有小酰基取代的唾液酸才能有效地与Ad37纤维蛋白结合, 12 (丙酰胺)几乎与唾液酸等效, 而13 (丁酰胺)和14 (戊酰胺)无结合能力, 因此推测酰胺取代基越大, 与结合位点的适配性越差; 但15 (苯甲酰胺)和16 (异丁酰胺)的结构与其他衍生物不同且亲和力较高, 因此推测口袋与酰胺之间存在其他结合方式。为了进一步研究N-酰基取代唾液酸的活性, 合成了经上述计算机模拟的一系列正酰基改性唾液酸及其多价HSA共轭物, 验证了计算机模拟的结果, 合成的一系列正酰基改性唾液酸中, 丙酰胺抑制活性最好, 并且显示多价丙酰胺唾液酸活性优于单价丙酰胺唾液酸, 但仍低于多价唾液酸, 所以唾液酸本身的乙酰胺基活性仍为最优。
由于功能化唾液酸的合成复杂, 另一种设计抑制剂的策略可能是用非碳水化合物唾液酸模拟物替代整个唾液酸核, 该模拟物具有病毒识别的关键属性, 与功能化唾液酸相比, 非碳水化合物唾液酸模拟唾液酸具有合成简单、官能团结构多样等优点。
|
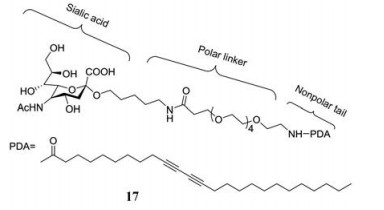
Aplander等[11]以唾液酸为基础合成了多价唾液酸偶联物, 其结构由3个不同的区域组成, 即唾液酸单元、极性连接区域以及嵌入10, 12-戊二十碳二烯酸(PDA)或脂质体的非极性尾巴。通过优化连接区域的长度和尾部, 得到以PDA为脂尾, 聚乙二醇(PEG4)为连接体的化合物17活性最好, EC50 = 4 μmol·L-1。并且发现配方和工艺会影响多价唾液酸偶联物的活性, 如果一个配方中唾液酸的总体密度过高, 受到空间位阻的影响, 唾液酸与纤维旋钮蛋白不能有效的结合, 所以在适宜的比例下唾液酸偶联物有较好的活性。之后证明唾液酸单位与PDA的比例为1:9, 以探测声波降解法合成的17的制剂是腺病毒的有效抑制剂, 在腺病毒结合抑制试验中EC50 = 0.9 μmol·L-1, 腺病毒感染抑制试验中EC50 = 0.7 μmol·L-1。因此, Aplander等发明了“分子湿巾” (molecular wipes)法, 可以用来治疗流行性角膜结膜炎(EKC)。
|
纤维旋钮蛋白的晶体结构表明, 3个已知的唾液酸结合位点之间的距离约为10 Å, 所以Spjut等[12]选择了3种小而灵活的柔性骨架, 设计合成了3个三价或四价唾液酸衍生物18 (ME0322)、19 (ME0323)及20 (ME0324), 希望较长的柔性连接可以使3个结合口袋被1个分子中的3个唾液酸残基同时占据。ME0322、ME0323和ME0324均以剂量依赖的方式抑制Ad37病毒粒子与HCE细胞的粘附, 其作用比唾液酸至少高两个数量级。其中最有效的化合物ME0322的活性与17价唾液酸-HAS偶联物相当, 比天然唾液酸单糖高4个数量级。之后Caraballo等[13]以18为先导化合物, 保留构成ME0322核心片段的三价氨基支架和与目标蛋白相互作用至关重要的唾液酸残基, 缩短了最初的连接基, 使核心片段更接近受体, 并引入异芳基环三唑基作为连接物, 与纤维旋钮建立新的接触, 设计并合成了一系列三唑连接基三价唾液酸衍生物。其中活性最好的是化合物21, 在Ad37对HCE细胞的结合抑制试验中, 其EC50值比先导化合物ME03222低3个数量级, 在Ad37对HCE细胞的感染抑制试验中, 活性也是先导化合物的140倍左右。并通过X射线衍射论证了化合物与Ad37纤维球状物之间存在一对一的结合模式, 然而, 连接基和核心片段的配体-受体相互作用未被观察到。
唾液酸衍生物主要用于治疗以唾液酸为受体的腺病毒亚型导致的眼部感染, 如流行性角膜结膜炎(EKC), 具有局部治疗可以避免系统治疗对碳水化合物类药物高清除率导致的疗效降低, 以及作为正常碳水化合物在细胞外水平发挥作用, 不良反应小的优点, 可作为以唾液酸为受体的腺病毒亚型的特异性抑制剂进行进一步的开发与研究。

|
腺病毒半胱氨酸蛋白酶负责处理病毒的衣壳和核心前体蛋白, 有助于腺病毒进入宿主细胞时病毒颗粒的脱衣壳, 它还参与细胞骨架蛋白的分裂导致宿主细胞的裂解, 是形成成熟的具有感染性的病毒颗粒所必不可少的酶, 且它没有人类同源物, 所以可作为腺病毒治疗的理想靶点。
腺病毒蛋白酶对多种蛋白酶抑制剂敏感, 包括木瓜蛋白酶抑制剂和绿茶儿茶素, 但这些抑制剂对腺病毒蛋白酶的特异性较低, 不适合作为治疗手段[14], 为了寻找高特异性腺病毒蛋白酶抑制剂, Pang等[15]对HIV病毒和丙型肝炎病毒的蛋白酶抑制剂进行了高通量筛选和计算机建模, 通过虚拟筛选发现化合物22 (2, 4, 5, 7-四硝基-9-芴酮)能够选择性且不可逆地抑制腺病毒半胱氨酸蛋白酶, 可作为一种新的先导化合物进一步优化。

|
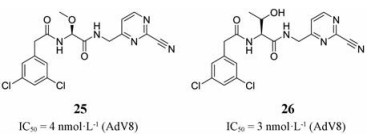
Eva Altmann课题组通过模拟腺病毒蛋白酶切割位点底物的多肽序列以及对选定的杂环腈进行重点筛选的双管齐下的快速发现方法确定了23 (四肽腈)与24 (嘧啶腈)两种腺病毒蛋白酶抑制剂作为结构优化的先导[16]。两种抑制剂均通过其腈基与腺病毒蛋白酶具有催化功能的Cys122残基共价结合, 此外四肽腈的P4苯环的3-氯代取代基与Ala46主链羰基氧之间存在卤键相互作用, 其所有酰胺基团都与蛋白酶形成氢键作用; 而嘧啶腈由于腈基被Cys122基团进攻而形成的硫酰亚胺基与Gln115形成氢键, 与四肽腈不同的是它不延伸到结合口袋中(图 4)。从四肽腈和嘧啶腈片段出发, 采用结构导向的药物化学策略对低纳摩尔级的抑制剂进行了优化。由于四肽腈渗透性差, 导致其体内活性远低于体外, 所以采用降低肽键数和分子量、提高渗透率的优化策略, 通过在P4苯基的2位上引入含有小极性或亲脂取代基和在P3中进行具有支链侧链的氨基酸的设计, 对P3和P4进行了修饰, 并将肽链与嘧啶腈结合, 用嘧啶核代替P1/P2酰胺键甘氨酸腈部分产生了高效的腺苷酸抑制剂, 具有高渗透性和良好的溶解性, 证明一个酰胺键的替换可大大提高其渗透性。其中嘧啶腈抑制剂25和26是一类具有类药性的先导化合物。
|

|
Figure 4 X-ray co-crystal structures: A, compound 23 bound in the catalytic site of AVP2 (PDB code: 4PIE), B, compound 24 bound in catalytic site of AVP2 (PDB code: 4PID). The pictures were made by Pymol |
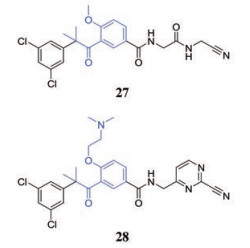
Eva Altmann课题组以四肽腈为先导分子, 设计了一种新型的类肽骨架, 以3, 5-二氯苯基作为P4部分, 以间位取代苯环替代四肽腈的NHCH(CH2Ph)作为P3部分, 并在苯环的邻位进行修饰, 得到了一类新型结构(图 5)。通过活性评价, 得到高效的腺病毒蛋白酶抑制剂27, IC50 = 0.04 μmol·L-1, 具有良好的渗透性, 但其溶解度较低。在进一步的优化中, 用4-氨基甲基-2-氰基嘧啶取代了P2-P1肽段, 最终得到了活性更好的化合物28, IC50 = 0.1 nmol·L-1, 具有高渗透性和良好的溶解度, 细胞病变效应(cytopathic effect, CPE)试验中EC50仅为3.9 μmol·L-1, 但细胞水平的抗病毒活性仍有所下降[17]。
|

|
Figure 5 Design of new scaffold based on tetrapeptide nitrile |
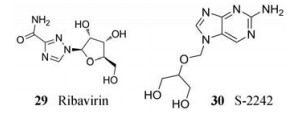
利巴韦林(1-β-D-磷酸核糖酯-1, 2, 4-三唑-3-甲酰胺, 29), 又名病毒唑, 是一种嘌呤核苷酸类似物, 具有体外抗腺病毒感染活性, 且对腺病毒不同亚型的活性有差异, 对C亚型的活性最强; 然而利巴韦林需达到的血浆浓度是其EC50的10倍。其作用机制可能是直接抑制病毒多聚酶, 以及通过免疫调节来耗竭三磷酸鸟苷、抑制RNA的加帽活动或增加新合成DNA的基因突变以间接抑制腺病毒感染, 其不良反应主要是引起温和可逆的镰状细胞贫血。利用利巴韦林治疗处于传播腺病毒高发期的患者, 效果不明显[18]。
3.1.2 S-2242S-2242 [2-氨基-7-(1, 3-二羟基-2-丙氧甲基)嘌呤, 30]是一种具有广谱抗DNA病毒活性的无环嘌呤衍生物, 细胞激酶磷酸化效率较高。S-2242具有细胞水平的抗腺病毒复制活性, 其活性和选择性(EC50 = 0.6 μmol·L-1, CC50 > 400 μmol·L-1)均优于西多福韦(HPMPC, EC50 = 2.0 μmol·L-1, CC50 = 83 μmol·L-1)与扎西他滨(ddC, EC50 = 1.4 μmol·L-1, CC50 = 231 μmol·L-1)[19]。
|
西多福韦属于无环膦酸核苷(acyclic nucleoside phosphonates, ANPs, 31), 是胞嘧啶核苷膦酰基甲醚衍生物[20], 可在宿主细胞酶的作用下生成活性代谢物西多福韦二磷酸酯, 通过竞争性抑制胞嘧啶, 整合到病毒DNA链中, 从而抑制病毒DNA的合成, 发挥广谱抗病毒活性, 是目前临床治疗腺病毒感染的首选药物。特别是针对腺病毒感染后早期治疗, 有效率很高。作为第一代ANPs类代表药物[21], 西多福韦结构中膦酸盐游离电荷的影响, 使得药物的生物利用度和细胞穿透性较低; 西多福韦作为人体阴离子转运体的底物, 它们会被动地聚集在肾小管, 使肾脏中的药物浓度高于其他组织从而引起严重的肾毒性, 极大地限制了其临床应用[22, 23]。
3.2.2 西多福韦前药将第一代ANPs结构中的磷酸基团制备成膦酸酯或膦酰胺前药[22], 其生物利用度与毒副作用往往得到极大改善。受此启发, 为克服西多福韦口服生物利用度低的问题, 人们合成了多种西多福韦亲脂前药。在腺病毒感染的细胞中, 一些前药的活性是西多福韦的5~300倍, 具有良好的选择性[23]。
环-西多福韦(cyclic-HPMPC, 32)是由西多福韦环合形成的内酯型前药, 化学性质稳定, 在细胞内能被磷酯酶催化转变成西多福韦发挥作用, 与西多福韦相比, 口服生物利用度和肾毒性均有改善[23]。但西多福韦和环-西多福韦均通过细胞的胞饮作用缓慢吸收, 因此, 它们的口服生物利用度有限。人们进而以西多福韦和环-西多福韦为母核, 设计一系列的长链脂肪基前药[24], 如化合物33~36 (十六烷丙氧基-环-西多福韦、十八烷乙氧基-环-西多福韦、十六烷丙氧基-西多福韦、十八烷乙氧基-西多福韦)等, 可以掩蔽磷酸基团, 明显提高药物的稳定性和脂溶性, 从而提高药物的口服生物利用度, 降低毒副作用[25]。
其中前药十六烷丙氧基西多福韦(又名brincidofovir), 具有可口服、抗病毒作用强和肾毒性低等优点, 在体内外对多种DNA病毒和逆转录病毒有强效抗病毒作用[26], Chimerix公司对此开展用于腺病毒和天花治疗的临床试验, 但brincidofovir抗CMV Ⅲ期临床试验失败, 其前药形式有待进一步优化。

|
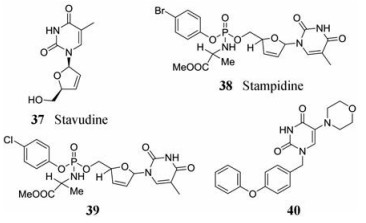
司他夫定(stavudine, d4T, 37)是一种嘧啶核苷类HIV逆转录酶抑制剂。它经过核苷和核苷酸激酶的作用在细胞内生成5'-三磷酸盐形式, 其中, 在核苷激酶活化下生成单磷酸盐是其限速步骤。为了克服司他夫定对细胞内核苷激酶活化的依赖, Uckun等[27]制备了一系列磷酸衍生物, 其中在抑制腺病毒诱导的斑块形成试验中化合物38 (stampidine)的EC50为0.017 μmol·L-1, 化合物39的EC50为0.027 μmol·L-1。这两种化合物在纳摩尔浓度下均有强效抗HIV活性, 然而, 这两种化合物都对其他DNA病毒(腺病毒除外)无效, 可用于治疗艾滋病患者中的腺病毒感染者。
3.3.2 5-氨基尿嘧啶类衍生物2018年, Nikitenko等[28]报道了1-(4-苯氧基苄基) 5-氨基尿嘧啶类人腺病毒抑制剂, 其中化合物40活性最好(EC50 = 0.5 μmol·L-1, CC50 = 47.6 μmol·L-1), 可作为先导化合物供进一步结构优化。
|
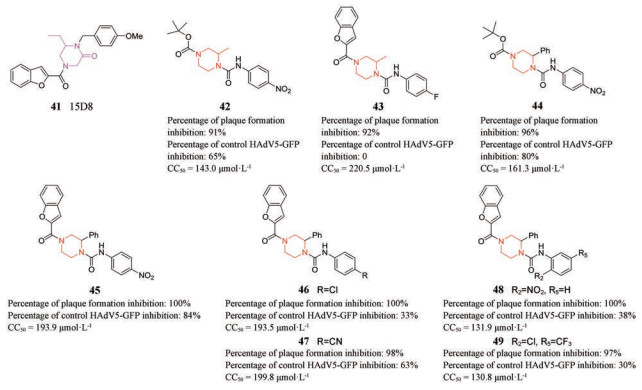
Sanchez-Cespedes等通过高通量筛选含有25 000多种小分子的化合物库, 发现了一种三取代哌嗪-2-酮苗头化合物41 (15D8)[29], 可在微摩尔浓度下显示出强大的抗腺病毒活性(表 1), 且细胞毒性低。作用机制可能是选择性地抑制腺病毒DNA在细胞核中的复制, 影响基因转录, 减少mRNA的生成, 以及影响病毒基因核定位。之后Sánchez-Céspedes等[30]基于前期化合物库的构效关系, 对先导化合物41进行结构修饰, 以提高其抗腺病毒活性, 先后得到三代取代哌嗪类腺病毒抑制剂, 并进行了腺病毒诱导空斑形成抑制试验以及细胞水平的腺病毒进入活性测试。其中用哌嗪-1环取代了41的哌嗪-2-酮-1环得到化合物42, 进一步修饰得到4-酰基-1-苯基氨基羰基-2-甲基哌嗪类衍生物43以及4-酰基-1-苯基氨基羰基-2-苯基哌嗪类衍生物44~49, 其中化合物44~49在抗病毒活性的菌斑试验中抑制率为96%~100% (表 2)。
| Table 1 EC50, CC50 and SI value of 41. EC50: Concentration of compound required to achieve 50% protection of cell cultures against adenovirus -induced cytotoxicity, CC50: Concentration required to reduce the viability of mock-infected cell cultures by 50%, SI: Selectivity index, the ratio of CC50/EC50 |
| Table 2 EC50, CC50 and SI value of 44-49 |
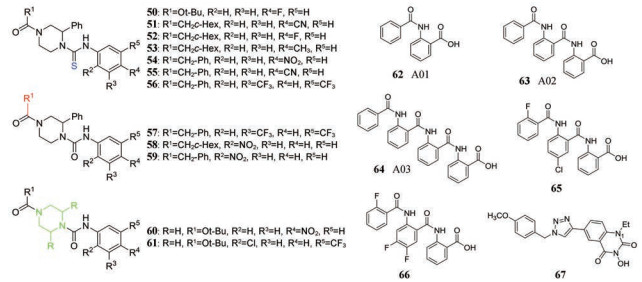
Mazzotta等[31]以4-酰基-1-苯基氨基羰基-2-苯基哌嗪类骨架的进一步全面修饰得到化合物50~61, 在纳摩尔和低微摩尔浓度下(EC50从0.6到5.1 μmol·L-1)抑制HAdV感染, 且有较低的细胞毒性, 机制研究发现这些衍生物在针对HAdV复制周期中转录、DNA复制以及随后的步骤发挥抗HAdV活性的方式存在差异, 其中58、59和61针对HAdV进入细胞后的早期步骤, 阻断HAdV基因组与细胞核的接触, 56抑制E1A基因的转录, 53、54、55和60干扰HAdV DNA复制, 50、51、52和57干扰HAdV DNA复制后的后续步骤未完全阐明的机制。由于这类化合物具有不同的作用机制, 可作为新颖的先导化合物供进一步研究。
|
|
Öberg等[32]开发了一种基于细胞的高通量筛选抗腺病毒化合物的方法, 其独特之处在于可以测量腺病毒11p型绿色荧光蛋白(GFP)表达载体(RCAd11pGFP)基因组在K562细胞系统中表达直接导致的荧光变化。该课题组利用该方法, 筛选了9 800种市售小分子, 其中化合物63 {A02, 2-[2-(苯甲酰氨基)苯甲酸]}是一种低毒、高效广谱抗腺病毒化合物, 它可以抑制几乎所有类型腺病毒的DNA复制(EC50 = 3.7 μmol·L-1, CC50 = 199 μmol·L-1)[33]。与之相比, 二聚体62 (A01)及四聚体64 (A03)均无抗腺病毒作用, 原因可能是62及64的大小与蛋白结合位点不匹配。在此基础上, 研究者又设计、合成并评价了42个新化合物, 其中最有效的是65与66, 其EC50分别为0.57和0.58 μmol·L-1。
4 机制不明的抑制剂化合物库的表型筛选是发现全新骨架分子的重要途经。本课题组运用铜(I)催化叠氮-炔环加成(CuAAC)反应建立了6-(1-苄基-1H-1, 2, 3-三唑-4-酰基)-3-羟基类喹唑啉-2, 4(1H, 3H)-二酮类化合物库, 经细胞水平的抗腺病毒活性筛选发现一批活性分子, 其中67活性最好(EC50 = 6.2 μmol·L-1, CC50 > 100 μmol·L-1), 优于阳性对照药物西多福韦(EC50 = 10 μmol·L-1, CC50 > 250 μmol·L-1)、扎西他滨(EC50 = 7.2 μmol·L-1, CC50 > 250 μmol·L-1)以及阿洛夫定(EC50 = 10 μmol·L-1, CC50 > 250 μmol·L-1)[34]。SARs研究表明, 与三唑相连的N1-R取代基和芳基上取代基的性质显著影响化合物的抗病毒活性, 与N1-甲基取代物相比, N1-乙基取代物具有更强的抗病毒活性; 在芳基上引入一个强吸电子基团可导致化合物失去抗腺病毒活性, 而引入给电子基则可以提高化合物的抗腺病毒活性, 但将芳基取代方式改为双取代后, 活性降低, 甚至完全丧失。尽管该类化合物抗腺病毒的精确作用机制与具体靶点仍有待鉴定, 但为新一代抗腺病毒药物提供了先导结构。
5 免疫调节剂 5.1 靶向HLA-A2的小分子抑制剂腺病毒蛋白E3-19K与人类白细胞抗原(HLA)的结合在腺病毒感染中起重要作用, HLA即人MHC I类分子, 参与区分体内的“自身”和“外来”蛋白, 介导T淋巴细胞裂解受感染细胞的免疫过程, 而腺病毒蛋白E3-19K与HLA结合, 将其保留在Ad感染细胞的内质网中, 从而阻断其向细胞表面逃逸, 破坏了HLA的免疫介导作用, 进而引起一系列胃肠、呼吸和眼病[35]。Ren等[36]通过虚拟筛选发现了Ad2 E3-19K和HLA相互作用抑制剂68~74, 在2.5~75 μmol·L-1微摩尔水平与HLA-A2结合。
5.2 病毒信号转导调控分子前苏联药物化学研究中心研制的盐酸阿比朵尔(75)是一种抗病毒药物和免疫刺激剂。石丽桥等[37]发现它对AdV-7具有体外抑制作用(EC50 = 29.9 μmol·L-1, CC50 = 166 μmol·L-1), 且呈现浓度依赖性。随后发现盐酸阿比朵尔对AdV-7无直接杀伤作用, 也不能阻止AdV-7的吸附和穿入, 其抗AdV-7的作用是在病毒进入细胞后的某个环节产生; 可能是因为腺病毒早期表达的E1A蛋白可影响干扰素介导的抗病毒反应的信号转导, 所以腺病毒对干扰素高度抵抗, 而盐酸阿比朵尔具有非特异性防御能力, 能够诱导产生干扰素, 对巨噬细胞有显著的活化作用[38]。

|
传统的新药研发是一个费时费力、成本昂贵和高风险的过程, 开发一种新药通常需要10~15年的时间, 药物研发投入也达到30亿美元。而老药新用是用现有的药物治疗一些新的疾病, 为现有药物寻找新适应症, 具有高效、低成本的特点, 研究人员平均只需要1~2年的时间来确定新的药物靶点, 研究成本也显著降低, 此外, 老药具有经证实的药物代谢动力学性质及安全性, 能很快进入Ⅱ期临床评估。因此近年来, “老药新用”策略日益受到重视和广泛应用, 特别是在抗病毒药物研发中, 老药正逐渐焕发生机。
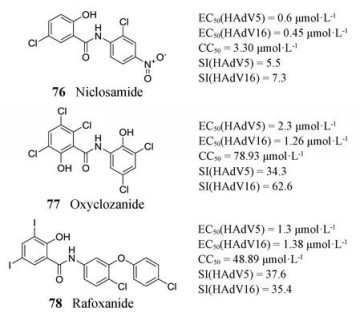
2009年, Javier Sánchez-Céspedes课题组筛选评估了三种核心为N-苯基苯甲酰胺的水杨酰苯胺类治疗绦虫感染的驱虫药氯硝柳胺(niclosamide, NIC, 76)、五氯柳胺(oxyclozanide, OXY, 77)和碘醚柳胺(rafoxanide, RAF, 78), 还对这些药物的细胞毒性进行了评估, 并对其作用机制进行了阐述。结果显示水杨酰苯胺类驱虫药在低微摩尔浓度下具有明显的抗腺病毒活性, 且细胞毒性较小。此外, 进一步研究表明, 此类药物发挥抗腺病毒活性的方式存在差异。氯硝柳胺和碘醚柳胺靶向腺病毒颗粒从核内体到核膜的转运, 而五氯柳胺则直接靶向腺病毒早期基因E1A转录[39]。所以水杨酰苯胺驱虫药适合进一步结构优化, 以开发新的抗病毒药物, 治疗免疫抑制患者和具有免疫能力社区获得性肺炎患者的腺病毒感染。
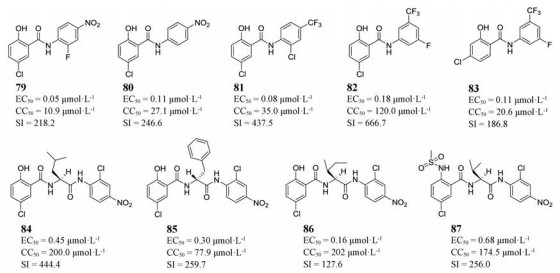
之后, 他们以前期筛选得到的氯硝柳胺为先导, 对其结构进行了优化, 得到了一系列的水杨酰胺衍生物。其中, 9个化合物(79~87)表现出从纳摩尔到亚微摩尔的EC50值和高选择性指数(SI > 100), 有比先导氯硝柳胺更好的抗腺病毒活性以及更广泛的安全窗口。进一步实验证明化合物80、86和87阻止腺病毒侵入过程, 79、82、83和84可能抑制DNA复制后的后续步骤, 而80和85则是抑制腺病毒的DNA复制[40]。
|
由于氯硝柳胺具有广泛的抗病毒活性, 所以一旦它们的抗病毒特性在体内得到验证, 这些衍生物不仅可以对腺病毒感染也为其他病毒感染的治疗具有较高的潜力。开发这些新型抗病毒药物的下一步将是进一步广泛评估它们在病毒感染动物模型中的有效性和安全性, 以及将它们推进到潜在的临床应用中。
7 总结与展望迄今为止还没有针对腺病毒感染的特异性药物批准上市, 对腺病毒感染通常采用广谱抗病毒治疗和免疫治疗, 临床上现有的非特异性抗腺病毒感染治疗方案在疗效或毒性方面的结果并不令人满意。通过本文对腺病毒以及抗腺病毒药物化学新进展的总结, 凝练当前具有广阔前景的抗腺病毒药物研发前沿方向, 例如多价态唾液酸类衍生物抑制剂、腺病毒半胱氨酸蛋白酶抑制剂、嘌呤及嘧啶类病毒复制抑制剂、哌嗪脲类抑制剂等。
|
目前, 抗腺病毒新药开发面临的主要问题是如何快速发现新的先导化合物, 基于高品质化合物库的高通量筛选并结合如基于片段的筛选、虚拟筛选或对筛选片段的进一步修饰优化等新理论、新方法是发现抗腺病毒先导物的重要途径。
对于已经发现的并确定靶点的先导化合物, 通过对其结合模式及构效关系的分析, 采用“优势结构”导向策略, 根据药物设计的基本原理如生物电子等排、骨架跃迁、分子杂合和前药修饰等来改变中心母核和侧链结构, 设计合成新的衍生物, 并通过系统的药代动力学、药效学和毒理学评价发现候选药物的方法, 依然是抗腺病毒药物的重要研究策略。
此外, 由于高效、研发成本小、安全可控性高的特点, 老药新用作为药物开发的新策略越来越受到重视, 并由此诞生了大量的新适应症药物。而水杨酰苯胺类化合物作为抗腺病毒药物的成功开发也证实了老药新用策略作为寻找新型抗腺病毒药物手段的可行性, 今后可以继续从临床药物或者具有活性数据的候选药物中筛选抗腺病毒先导物。不容忽视的是, 传统中药具有多靶点作用机制、不易产生耐药性等优点, 从中药中筛选高效低毒的抗腺病毒分子是今后的研究热点之一[41-44]。
同时, 对腺病毒的认识还需进一步深入, 譬如:在病毒脱衣壳中触发其进行结构变化的精确位置、病毒DNA双链与核心蛋白V、Ⅶ、Mu的结构信息, 是否有特异性受体或其他受体, 腺病毒进入细胞的信号通路以及病毒与免疫之间的关系等, 这有助于指导优化临床抗腺病毒疗法和研发新的抗腺病毒药物和疫苗。
| [1] |
Lenaerts L, De Clercq E, Naesens L. Clinical features and treatment of adenovirus infections[J]. Rev Med Virol, 2008, 18: 357-374. |
| [2] |
Wu L, Zhou P, Ge X, et al. Deep RNA sequencing reveals complex transcriptional landscape of a bat adenovirus[J]. J Virol, 2013, 87: 503-511. DOI:10.1128/JVI.02332-12 |
| [3] |
Gao WJ, Jin Y, Duan ZJ. Research progress in human adenovirus[J]. Chin J Virol (病毒学报), 2014, 30: 193-200. |
| [4] |
Vassal-Stermann E, Mottet M, Ducournau C, et al. Mapping of adenovirus of serotype 3 fibre interaction to desmoglein 2 revealed a novel 'non-classical' mechanism of viral receptor engagement[J]. Sci Rep, 2018, 8: 8381. DOI:10.1038/s41598-018-26871-x |
| [5] |
Cheng L, Huang X, Li X, et al. Cryo-EM structures of two bovine adenovirus type 3 intermediates[J]. Virology, 2014, 450-451: 174-181. |
| [6] |
Chen NN, Xiang DX, Zheng CL. Adenovirus and its research progress[J]. J Dalian Med Univ (大连医科大学学报), 2010, 32: 586-590. |
| [7] |
Johansson S, Nilsson E, Qian W, et al. Design, synthesis, and evaluation of N-acyl modified sialic acids as inhibitors of adenoviruses causing epidemic keratoconjunctivitis[J]. J Med Chem, 2009, 52: 3666-3678. |
| [8] |
Kaneko H, Mori S, Shigeta S, et al. Antiviral effect of sulfated sialyl lipid against a clinical strain of adenovirus[J]. Nippon Ganka Gakkai Zasshi, 2003, 107: 196-201. |
| [9] |
Johansson SM, Arnberg N, Elofsson M, et al. Multivalent HSA conjugates of 3′-sialyllactose are potent inhibitors of adenoviral cell attachment and infection[J]. Chembiochem, 2005, 6: 358-364. DOI:10.1002/cbic.200400227 |
| [10] |
Johansson SM, Nilsson EC, Elofsson M, et al. Multivalent sialic acid conjugates inhibit adenovirus type 37 from binding to and infecting human corneal epithelial cells[J]. Antiviral Res, 2007, 73: 92-100. DOI:10.1016/j.antiviral.2006.08.004 |
| [11] |
Aplander K, Marttila M, Manner S, et al. Molecular wipes: application to epidemic keratoconjuctivitis[J]. J Med Chem, 2011, 54: 6670-6675. DOI:10.1021/jm200545m |
| [12] |
Spjut S, Qian W, Bauer J, et al. A potent trivalent sialic acid inhibitor of adenovirus type 37 infection of human corneal cells[J]. Angew Chem Int Ed, 2011, 50: 6519-6521. DOI:10.1002/anie.201101559 |
| [13] |
Caraballo R, Saleeb M, Bauer J, et al. Triazole linker-based trivalent sialic acid inhibitors of adenovirus type 37 infection of human corneal epithelial cells[J]. Org Biomol Chem, 2015, 13: 9194-9205. DOI:10.1039/C5OB01025J |
| [14] |
Lenaerts L, Naesens L. Antiviral therapy for adenovirus infections[J]. Antiviral Res, 2006, 71: 172-180. DOI:10.1016/j.antiviral.2006.04.007 |
| [15] |
Pang YP, Xu K, Kollmeyer TM, et al. Discovery of a new inhibitor lead of adenovirus proteinase: steps toward selective, irreversible inhibitors of cysteine proteinases[J]. FEBS Lett, 2001, 502: 93-97. DOI:10.1016/S0014-5793(01)02672-2 |
| [16] |
Mac SA, Grosche P, Ellis D, et al. Discovery and structure-based optimization of adenain inhibitors[J]. ACS Med Chem Lett, 2014, 5: 937-941. DOI:10.1021/ml500224t |
| [17] |
Grosche P, Sirockin F, Mac Sweeney A, et al. Structure-based design and optimization of potent inhibitors of the adenoviral protease[J]. Bioorg Med Chem Lett, 2015, 25: 438-443. DOI:10.1016/j.bmcl.2014.12.057 |
| [18] |
Wang H, Wu QP. Studies in anti-virus nucleoside analogues[J]. Life Sci Instrum (生命科学仪器), 2006, 4: 11-14. |
| [19] |
Naesens L, Lenaerts L, Andrei G, et al. Antiadenovirus activities of several classes of nucleoside and nucleotide analogues[J]. Antimicrob Agents Chemother, 2005, 49: 1010-1016. DOI:10.1128/AAC.49.3.1010-1016.2005 |
| [20] |
Yi H, Li ZR. Cidofovir - an antiviral agent[J]. Food Drug (食品与药品), 2005, 7: 15-18. |
| [21] |
De Clercq E. Cidofovir in the therapy and short-term prophylaxis of poxvirus infections[J]. Trends Pharmacol Sci, 2002, 23: 456-458. DOI:10.1016/S0165-6147(02)02091-6 |
| [22] |
Andrei G, Topalis D, Schutter TD, et al. Insights into the mechanism of action of cidofovir and other acyclic nucleoside phosphonates against polyoma- and papillomaviruses and non-viral induced neoplasia[J]. Antiviral Res, 2015, 114: 21-46. DOI:10.1016/j.antiviral.2014.10.012 |
| [23] |
Fang SS, Zhang S, Cao GY. Advances in the study of the nucleoside antiviral drug cidofovir[J]. China Pharm (中国药房), 2016, 21: 3022-3024. |
| [24] |
Beadle JR, Hartline C, Aldern KA, et al. Alkoxyalkyl esters of cidofovir and cyclic cidofovir exhibit multiple-log enhancement of antiviral activity against cytomegalovirus and herpesvirus replication in vitro[J]. Antimicrob Agents Chemother, 2002, 46: 2381-2386. DOI:10.1128/AAC.46.8.2381-2386.2002 |
| [25] |
Gao MY, Liu HT, Lian YF, et al. Research progress in prodrugs of acyclic nucleoside phosphonates antiviral agents[J]. Chin J Org Chem (有机化学), 2019, 39: 974-981. DOI:10.6023/cjoc201808018 |
| [26] |
Hostetler KY. Synthesis and early development of hexadecyloxypropylcidofovir: an oral antipoxvirus nucleoside phosphonate[J]. Viruses, 2010, 2: 2213-2225. DOI:10.3390/v2102213 |
| [27] |
Uckun FM, Pendergrass S, Qazi S, et al. Phenyl phosphoramidate derivatives of stavudine as anti-HIV agents with potent and selective in-vitro antiviral activity against adenovirus[J]. Cheminform, 2004, 39: 225-234. |
| [28] |
Nikitenko NA, Gureeva ES, Ozerov AA, et al. 1-(4-Phenoxybenzyl) 5-aminouracil derivatives and their analogues-novel inhibitors of human adenovirus replication[J]. Acta Nat, 2018, 10: 58-64. DOI:10.32607/20758251-2018-10-2-58-64 |
| [29] |
Sanchez-Cespedes J, Moyer CL, Whitby LR, et al. Inhibition of adenovirus replication by a trisubstituted piperazin-2-one derivative[J]. Antiviral Res, 2014, 108: 65-73. DOI:10.1016/j.antiviral.2014.05.010 |
| [30] |
Sánchez-Céspedes J, Martínez-Aguado P, Vega-Holm M, et al. Martínez-Aguado P, Vega-Holm M, et al. New 4-acyl-1-phenylaminocarbonyl-2-phenylpiperazine derivatives as potential inhibitors of adenovirus infection. Synthesis, biological evaluation and structure-activity relationships[J]. J Med Chem, 2016, 59: 5432-5448. DOI:10.1021/acs.jmedchem.6b00300 |
| [31] |
Mazzotta S, Marrugal-Lorenzo JA, Vega-Holm M, et al. Optimization of piperazine-derived ureas privileged structures for effective antiadenovirus agents[J]. Eur J Med Chem, 2020, 185: 111840. DOI:10.1016/j.ejmech.2019.111840 |
| [32] |
Öberg CT, Strand M, Andersson EK, et al. Synthesis, biological evaluation, and structure-activity relationships of 2-[2-(benzoylamino)benzoylamino]benzoic acid analogues as inhibitors of adenovirus replication[J]. J Med Chem, 2012, 55: 3170-3181. DOI:10.1021/jm201636v |
| [33] |
Andersson EK, Strand M, Edlund K, et al. Small-molecule screening using a whole-cell viral replication reporter gene assay identifies 2-{[2-(benzoylamino)benzoyl]amino}-benzoic acid as a novel antiadenoviral compound[J]. Antimicrob Agents Chemother, 2010, 54: 3871-3877. DOI:10.1128/AAC.00203-10 |
| [34] |
Kang D, Zhang H, Zhou Z, et al. First discovery of novel 3-hydroxy-quinazoline-2, 4(1H, 3H)-diones as specific anti-vaccinia and adenovirus agents via 'privileged scaffold' refining approach[J]. Bioorg Med Chem Lett, 2016, 26: 5182-5186. DOI:10.1016/j.bmcl.2016.09.071 |
| [35] |
Li L, Santarsiero BD, Bouvier MJ. Structure of the adenovirus type 4 (species E) E3-19K/HLA-A2 complex reveals species-specific features in MHC class Ⅰ recognition[J]. J Immunol, 2016, 197: 1399-1407. DOI:10.4049/jimmunol.1600541 |
| [36] |
Ren J, Dsouza NR, Deng H, et al. Discovery of small molecule inhibitors of adenovirus by disrupting E3-19K/HLA-A2 interactions[J]. Bioorg Med Chem Lett, 2018, 28: 2837-2841. DOI:10.1016/j.bmcl.2018.07.036 |
| [37] |
Shi LQ, Yang ZQ, He J, et al. Antiviral effect of arbidol hydrochloride on adenovirus type 7 in vitro[J]. Med J Wuhan Univ (武汉大学学报医学版), 2006, 27: 66-68. |
| [38] |
Sharma P, Martis PC, KJDA Excoffon. Adenovirus transduction: more complicated than receptor expression[J]. Virology, 2017, 502: 144-151. DOI:10.1016/j.virol.2016.12.020 |
| [39] |
Marrugal-Lorenzo JA, Serna-Gallego A, Berastegui-Cabrera J, et al. Repositioning salicylanilide anthelmintic drugs to treat adenovirus infections[J]. Sci Rep, 2019, 9: 17. DOI:10.1038/s41598-018-37290-3 |
| [40] |
Xu JM, Berastegui-Cabrera J, Chen HY, et al. Structure-activity relationship studies on diversified salicylamide derivatives as potent inhibitors of human adenovirus infection[J]. J Med Chem, 2020, 63: 3142-3160. DOI:10.1021/acs.jmedchem.9b01950 |
| [41] |
Shang L, Qu ZY, Ning LL, et al. The anti-adenovirus effect of astragaloside Ⅳ in vitro[J]. Chin Pharmacol Bull (中国药理学通报), 2013, 29: 854-858. |
| [42] |
Yang Y, Peng X, Zhu N, et al. Antiviral activities of flavonoids extracted from the core of lichi chinensis against adenovirus in vitro[J]. Med J Wuhan Univ (武汉大学学报医学版), 2014, 35: 41-45. |
| [43] |
Karimi A, Moradi MT, Alidadi S, et al. Anti-adenovirus activity, antioxidant potential, and phenolic content of black tea (Camellia sinensis Kuntze) extract[J]. J Complement Integr Med, 2016, 13: 357-363. |
| [44] |
Shang L, Qu ZY, Wei FX, et al. Advances in studies on anti-adenovirus of Chinese materia medica and its active components[J]. Chin Tradit Herb Drugs (中草药), 2012, 43: 2071-2076. |