 2020, Vol. 55
2020, Vol. 55



病毒性传染病始终严重威胁着人类的生命健康。一些原有的病毒性疾病包括艾滋病、乙肝、流感等疾病还未彻底根除, 每年可造成数百万人死亡。而一些新暴发的病毒性疾病也开始席卷全球, 包括寨卡病毒传染病、基孔肯雅热病等正从疫情高发区逐渐向其他国家及地区扩散。面对许多病毒性疾病无药治疗或者尚未根除的问题, 早日研制出小分子药物是最有效的解决手段。天然产物具有来源广泛、结构独特等特点, 而我国的天然产物自然资源丰富, 特别是对中草药的利用历史悠久, 在天然药物领域有着得天独厚的优势。本文精选了近十年的文献, 总结了国内外天然产物抗病毒研究的新进展。
1 抗艾滋病病毒(HIV/AIDS)艾滋病(acquired immune deficiency syndrome, AIDS)是由人免疫缺陷病毒(human immunodeficiency virus, HIV)引起的一种破坏人体免疫系统的慢性传染病。病毒是由脂层双膜、球形基质以及蛋白p24形成的半锥形衣壳组成。衣壳内含有病毒的RNA和酶, 包括逆转录酶(reverse transcriptase, RT)、整合酶(integrase, IN)、蛋白酶(protease, PR)以及核心壳蛋白(nucleocapsid protein, NCp)。HIV-1感染宿主细胞主要分为:吸附与融合、逆转录、整合、转录、翻译、组装、出芽及成熟等过程(图 1)[1]。理论上, 阻断病毒复制周期的任何一个环节, 都可以实现抗病毒的目的。

|
Figure 1 The life cycle of HIV-1[1] |
萜类化合物是分布在动植物界特别是在植物香精油(essential oil)中的挥发性物质, 是重要的天然药物化学成分。萜类化合物主要分为单萜、倍半萜、二萜、三萜化合物等。
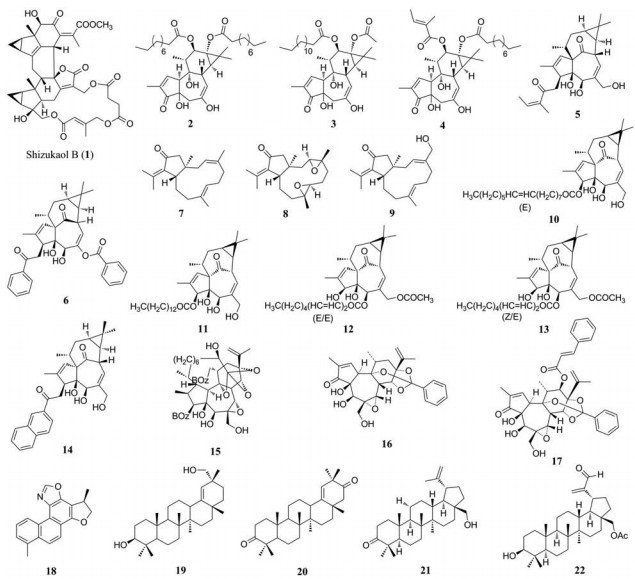
Fang等[2]从银线草中分离得到的乌药烷二聚倍半萜类化合物, 即银线草醇B (shizukaol B, 1), 其对野生型和突变型(K103N、Y181C) HIV-1均显示出亚微摩尔的抑制活性, EC50值分别为0.22、0.47、0.50 μmol·L-1, 但在C8166细胞系中有着较强的细胞毒性(CC50 = 20 nmol·L-1)。研究证明该类化合物可特异性抑制HIV-1核糖核酸酶H (RNase H)的活性, 从而抑制逆转录酶活性, 但对DNA聚合酶活性无影响, 是新结构类型的RNase H抑制剂[3]。
二萜类化合物广泛分布于自然界中, 约有120种骨架, 常见的主要是贝壳杉烷、赤霉烷、阿替烷、乌头烷等。Nothias-Scaglia等[4]评估了一系列天然二萜化合物(包括daphnane、tigliane和jatrophane二萜类化合物)的抗HIV和抗基孔肯雅病毒活性, 结果表明tigliane型二萜酯(2~4)和ingenane型二萜酯(5、6)在体外以纳摩尔水平抑制HIV复制。研究人员通过分析抗病毒数据证实这类天然产物在抑制两种病毒之间存在着线性关系, 猜测它们是通过与富含半胱氨酸的C1结构域的相互作用来调节蛋白激酶C同工酶(PKC)的活性。
Pardo-Vargas等[5]从加勒比海的octocoral Eunicea laciniata分离出一对dolabellane型二萜化合物(7)的对映异构体, 具有弱抗HIV活性和低毒性(EC50 = 190 μmol·L-1, CC50 = 2 570 μmol·L-1), 对其进行氧化, 衍生物8和9的抗HIV活性提高了100倍(EC50 = 0.73 μmol·L-1, 0.69 μmol·L-1; CC50 = 1 690 μmol·L-1, 980 μmol·L-1; SI = 2 315, 1 420), 而且相较于奈韦拉平(nevirapine), 其细胞毒性更低, 选择性更高(EC50 = 0.25 μmol·L-1, CC50 = 320 μmol·L-1, SI = 1 280)。构效关系(SAR)表明, dolabellane骨架中母环的绝对构型似乎并不影响生物活性。
Huang等[6]从Euphorbia ebracteolata的根中分离出多个ingenane型二萜化合物, 化合物(10~13)的IC50值在0.7~9.7 nmol·L-1之间, 选择性较高(SI > 10 000)。SAR研究发现, 在C-3、C-5或者C-20位上有长脂肪链取代基的化合物具有较强的抗病毒活性, 在C-20处酯化的化合物有较低的选择性。此外, Liu等[7]对大戟科植物Euphorbia kansui的甲醇提取物分离得到ingenane型酯, 化合物(14)的抗HIV-1活性最强(EC50 = 1.3 nmol·L-1)。另有研究表明, ingenane型二萜类化合物在体外和体内均可以有效地重新激活潜伏的HIV, 且细胞毒性较低[8, 9]。
Daphnane型二萜化合物—gnidimacrin (15)以前被报道过具有强效抗癌活性, Huang等[10]发现该化合物能激活病毒潜伏并以皮摩尔浓度杀死持续感染的细胞, 还以低于10 pmol·L-1的平均浓度抑制HIV-1 R5病毒感染外周血单核细胞(PBMCs)。研究人员推测其抗病毒活性与病毒辅助受体CCR5的下调有关。Vidal等[11]基于HPLC的活性谱分析, 从Daphne gnidium的地上部分的二氯甲烷提取物中也得到4种daphnane型二萜的衍生物, 化合物16、17通过直接干扰细胞表面的两种HIV受体CCR5和CXCR4的表达发挥较强的抗逆转录病毒活性, 且无细胞毒性。
Zhang等[12]从丹参的细胞培养物中得到了4种新的含有噁唑的二萜化合物。在HIV-1抑制试验中, 化合物18以剂量依赖性的方式抑制Tat介导的LTR转录活性, 作为HIV-1的转录抑制剂, 在亚微摩尔水平上阻止病毒复制。迄今为止, 尚无靶向HIV转录过程的抗逆转录病毒药物。因此, 该研究可能提供了一种有希望的新先导化合物。
Bazzocchi课题组从Cassine xylocarpa和Maytenus jelskii中分离得到多种新型齐墩果-18-烯三萜类化合物(19~22), 研究证明它们是增强子依赖性转录的抑制剂。该类化合物的总氧化水平、区域取代模式和官能团类型与抗病毒活性密切相关, 含有两个氧化位置的化合物比单氧化的化合物具有更高的抑制活性; C-3位被羟基取代是有利的, 而乙酰化是不利的[13, 14]。
|
苯丙素类是指基本母核有一个或几个C6-C3单元的天然有机化合物类群, 它们广泛存在于中药中, 主要包括简单苯丙素类、香豆素类、木质素类。
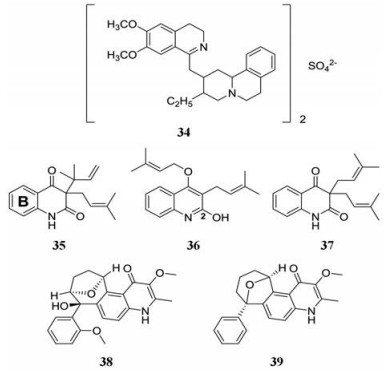
研究发现, 香豆素类分子可以靶向HIV-1 RT, 抑制聚合酶活性, 而且还能抑制HIV-1 PR和IN[15]。Esposito等[16]以香豆素为骨架, 针对IN和RNase H的活性位点进行修饰, 从而实现对病毒多靶点的抑制。大多数合成的香豆素衍生物能很好地结合到两个活性位点中, 其中活性最突出的衍生物23对IN和RNase H的抑制活性可达微摩尔水平, IC50值分别为6.75 ± 0.51和6.45 ± 0.45 μmol·L-1。
Sonar等[17]从圣罗勒叶的二氯甲烷提取物中分离得到十四烷基阿魏酸酯(24), 可以抑制HIV-1的RNase H活性, 对其进行结构修饰得到系列衍生物, N-油烯基咖啡酰胺衍生物(25)对RNase H和聚合酶均显示出极强的抑制活性, 分子模拟和Yonetani-Theorell分析发现它不仅结合NNRTIs结合口袋附近的变构位点, 也能结合RNase H的催化位点(图 2)。

|
Figure 2 Putative binding mode of 25: (a) 2D representation of 25 and binding pocket 1 interacting residues; (b) 2D representation of 25 and binding pocket 2 interacting residues |
(+)-Calanolide A (26)是第一种被发现的天然产物类HIV-1逆转录酶抑制剂[18], 其可以结合两个以上的活性位点来抑制逆转录酶。这类天然产物可以抑制对核苷类和非核苷类逆转录酶抑制剂产生抗性的多种HIV-1突变株。在26的衍生物中, 27显示出对病毒野生株和突变株Y181C极强的抑制活性(EC50值分别为7.4和0.46 nmol·L-1)和较高的选择性(SI = 1 417)[19]。
Dong等[20]基于HIV-1蛋白酶三维结构以及计算机辅助药物设计, 设计合成了一批四环双吡喃香豆素及其类似物, 化合物28能够抑制HIV-1逆转录酶和蛋白酶, IC50值分别为3.56和0.78 μmol·L-1, 28具有全新结构, 可作为新的先导化合物。
Zhang等[21]通过生物测定Justicia gendarussa的根和叶的提取物, 分离得到具有抗HIV-1活性的芳基萘木脂素糖苷patentiflorin A (29)和justiprocumin B (30), 对化合物29进一步结构修饰时发现喹啉吡喃氧基是抗HIV活性所必需的结构。化合物30显示出对广谱HIV病毒株的有效活性, EC50值在15~21 nmol·L-1内, 并且还显示出对多种抗性突变株的强效抑制活性。这类化合物可能是作用于HIV-1复制周期中的后期, 但其机制需要进一步的研究[21, 22]。

|
酚酸类化合物是一类含有酚环的有机酸, 一般作为次生代谢产物广泛存在于自然界中。姜黄素(31)能选择性地抑制HIV长末端基因复制的表达, 从而有效地阻止HIV的急性和慢性感染。研究证明, 在HIV复制周期中, 31还能抑制HIV-1 PR和IN的功能, IC50值分别为40和100 μmol·L-1[23]。
Tat蛋白在HIV-1复制周期和病毒感染的发病机制中起关键作用。研究证实用Tatve质粒转染后, 白藜芦醇(32)以浓度依赖性方式增加细胞内NAD+水平和SIRT1蛋白表达, 从而减弱Tat介导的HIV-1 LTR反式激活, 抑制病毒复制[24]。
多种间苯三酚类天然产物具有抗HIV活性[25], Chauthe等[26]基于植物来源的间苯三酚, 通过改变芳环和亚甲基桥上的取代合成了多种二聚间苯三酚。化合物33在病毒分离株感染的人CD4+ T细胞系(CEM-GFP)中显示出比齐多夫定(AZT, EC50 = 1.05 ± 0.07 μmol·L-1)更好的抑制活性, EC50值为0.28 μmol·L-1, 但是, 33对转录酶的抑制非常弱, 表明它可能涉及另外的机制。
Kamng'ona等[27]在非洲植物Myrothamnus flabellifolia中分离得到3, 4, 5-三氧没食子基奎宁酸, 与逆转录酶的结合动力学实验表明其为非竞争性抑制剂, IC50值为34 μmol·L-1。
1.4 生物碱类化合物生物碱是一类重要的碱性含氮天然有机化合物, 在动植物中的含量较丰富。来自于Cephaclis pecacuanha的O-甲基吐根碱(34)可以与酶-模板-启动子-底物复合物结合来抑制HIV RT聚合反应的延长, 从而发挥抗HIV作用。近期, Chaves Valadão等[28]的研究发现34对GHOST细胞(EC50 = 0.1 mmol·L-1)和外周血单核细胞(PBMC)中的HIV-1野生株和突变株M184V均表现出抑制作用(EC50值在0.012~0.03 μmol·L-1之间)。

|
喹诺酮生物碱(35、36)在CEM-GFP细胞系中表现出抗HIV活性(EC50值分别为2.99和3.80 μmol·L-1)[29], Ahmed等[30]以35为先导化合物, 设计合成了45种喹啉2, 4-二醇的烷基化衍生物, SAR研究表明未取代的环B和游离的2-OH基团对于抗HIV活性是必需的, 并且C-3位上还需要异戊二烯基。其中, 化合37 (EC50 = 2.35 μmol·L-1)与先导化合物相比更有效, 治疗指数(TI = 26.64)与AZT (TI = 23.07)相当。
Melochia odorata的甲醇提取物中分离得到3种4-喹诺酮类生物碱, 化合物38、39在体外感染的CEM-TART细胞中显示出对HIV-1的显著细胞保护作用, 但39的毒性要低于38。SAR结果表明C-3中的甲氧基和喹诺酮环并苯环是必不可少的结构, 而且双环系统的醚桥头从C-10向C-9的转变略微增强了抗HIV活性并降低了毒性[31]。
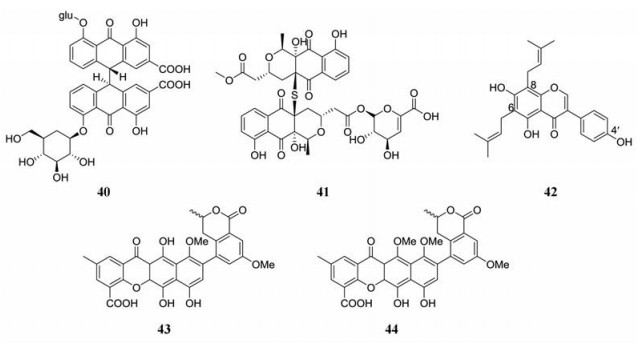
1.5 其他类化合物Esposito等[32]成功地在大黄中分离出醌类化学成分sennoside A (40), 其在低微摩尔范围内抑制HIV-1逆转录酶、整合酶的功能从而抑制病毒复制, 代表了一种双结合位点抑制剂的新型骨架, 但需要进一步化学修饰以提高活性。
|
He等[33]在链霉菌属KIB3133中分离出8种新的硫桥联吡喃萘醌(PNQ)二聚体, naquihexcin E (41)表现出中等的抗HIV活性(EC50 = 2.8 μmol·L-1)。
Lee等[34]从刺桐属植物Senegalensis中分离提取得到8个异戊烯异黄酮化合物, 能够抑制HIV-1蛋白酶的活性, IC50值在0.5~30.0 μmol·L-1之间, 化合物42对病毒的抑制活性最强。SAR研究发现6和8位的2个异戊烯基与4'位羟基与活性相关。Omolo等[35]发现来自Pyrenacantha kaurabassana Baill块茎的两种新的黄酮(43、44)属于侵入抑制剂, 显示中等抗HIV活性(EC50值分别为21和2 μg·mL-1)。
2 抗乙肝病毒(hepatitis B virus, HBV)乙肝病毒是一个大球形颗粒, 其外层是磷脂双分子层, 脂膜上镶嵌着大、中、小3种包膜蛋白, 即乙肝病毒表面抗原(hepatitis B surface antigen, HBsAg); 内层是一个正二十面体的核壳体, 由核心抗原(hepatitis B core antigen, HBcAg)聚集形成。HBV完整的生命周期包括:侵入、脱壳、释放遗传物质、合成cccDNA、转录、翻译、核壳体的包装、逆转录、DNA复制、表面蛋白包裹修饰、分泌出胞等过程。
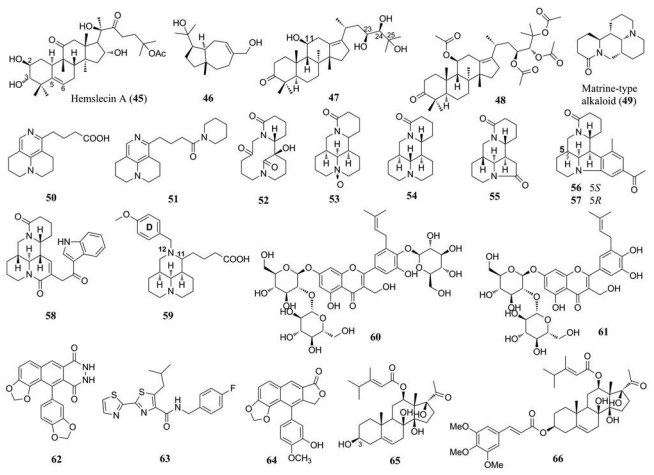
2.1 萜类化合物Hemslecin A (45), 一种广泛存在的葫芦烷萜类, 处于炎性疾病的临床实验中。Guo等[36]评估了45及其衍生物在HepG2.2.15细胞中的抗HBV活性, 发现45对HBV DNA复制具有中等抑制活性(EC50 = 11.4 μmol·L-1), 同时也具有明显的毒性(CC50 = 66.2 μmol·L-1), 导致SI值较低。SAR研究发现C-2/C-3上的羟基发生单酰化或者双酰化都会增强抗病毒活性, 并且会降低细胞毒性; C-5 (6)处的双键发生环氧化之后, 在保持抑制病毒活性(EC50 = 11.6 μmol·L-1)的同时, 也可以降低细胞毒性(CC50 > 1745 μmol·L-1)。
Ma等[37]从五味子(Schisandra wilsoniana)的果实中分离出3种胡萝卜素型倍半萜类化合物, 在浓度为50 μg·mL-1时, 化合物46显示出抗病毒的活性, 分别以76.5%和28.9%的抑制率抑制HBsAg和E抗原(HBeAg)分泌。
Zhang等[38, 39]合成了三萜类化合物泽泻醇A (alisol A, 47)的酯衍生物, SAR研究显示, 乙酰化C-25位上的羟基是维持抗HBV活性所必要的, 在C-11、C-23、C-24位上引入体积小的酰氧取代基增加抗HBV活性。其中, 化合物48对HBsAg和HBeAg的分泌具有高抑制活性, EC50值分别为0.004 8和0.011 mmol·L-1, 选择性指数(SIHBsAg > 333, SIHBeAg > 145)也十分突出。
|
苦参碱类生物碱抗肝炎病毒, 已被广泛应用于乙型肝炎的临床治疗。Zhang等[40]从苦参(Sophora flavescens)的根中分离出几种苦参碱类生物碱(49)及其类似物。体外抗病毒活性研究表明, 所有的生物碱均表现出有效的抗病毒活性, 在0.2 mmol·L-1或者0.4 mmol·L-1的非细胞毒性浓度下, 化合物50~54分别以37.2%~46.0%的抑制率抑制HBsAg的分泌, 具有与苦参碱相似的活性(浓度为0.4 mmol·L-1时为抑制率34.7%)。
他们还从苦豆子(Sophora alopecuroides)的种子中分离出4种新的苦参碱类生物碱。化合物55具有前所未有的6/5/6/6环系, 而化合物56和57是一对具有不寻常骨架的苦参碱-苯乙酮生物碱立体异构体, 化合物58具有吲哚嗪取代基, 在0.2 mmol·L-1或者0.4 mmol·L-1的非细胞毒性浓度下, 抑制率均大于50%, 比拉米夫定(在浓度为1.0 mmol·L-1时, 抑制率为31.5%)更有效[41, 42]。
氧化苦参碱(53)作用机制与核苷不用, 其通过下调宿主热应激同源物70 (Hsc70)表达。由于Hsc70不是病毒酶, 该类抑制剂不仅抑制野生型HBV的复制, 而且还抑制耐药株的复制[43]。Du等[44]设计并合成了26种新型N-取代的苦参碱类似物, 评价其对Hsc70 mRNA表达的抑制作用。SAR结果显示, C-11位的羧基是活性所必需的; 苦参碱的D环可能并非活性所需; 在D环开环后的骨架中, 12位氮原子上引入取代基, 尤其是取代的苄基(59), 可显著改善活性。
2.3 其他类化合物Wan等[45]从Ophioglossum pedunculosum中提取得到7种新的类黄酮葡萄糖苷, 化合物60、61显示出对HBsAg分泌具有剂量依赖性的抑制作用, 但两者的活性方面存在着较大差距。结构差异主要体现在葡萄糖基的数量不同, 与抗HBV活性相关。
Ying等[46]发现helioxanthin的类似物62表现出有效的抗HBV活性(EC50 = 0.08 ± 0.02 μmol·L-1), 且细胞毒性较小。研究发现它在HBV感染细胞中, 通过降低肝细胞核因子4 (HNF-4)、HNF-3和胎蛋白因子与前核心/核心启动子增强子Ⅱ区域的结合来抑制所有HBV启动子的活性, 从而阻断病毒基因表达和复制。
Yang等[47]发现来源于天然产物leucamide A的双杂环串联衍生物isothiafludine (NZ-4, 63)能够抑制HepG2.2.15细胞内HBV复制, EC50值为1.33 μmol·L-1, 并且还抑制各种耐药突变株的复制。机制研究发现63是通过干扰衣壳组装过程中HBV前基因组RNA (pgRNA)和核心抗原(HBcAg)之间的相互作用来抑制HBV DNA的复制。
Janmanchi等[48]合成了一系列helioxanthin (芳基萘木酯素内酯)类似物并评估了它们的抗HBV活性。化合物64显示出最有效的活性, 抑制HepA2细胞中HBsAg和HBeAg的分泌, EC50分别为0.06和0.14 μmol·L-1。机制研究发现, 64不仅抑制病毒DNA复制, 而且还抑制mRNA、核心蛋白和病毒启动子的活性[49]。
从白首乌中提取的caudatin (65)通过抑制细胞增殖和诱导细胞凋亡去除原发性肝癌。研究人员发现65可以抑制HBsAg分泌和DNA复制, EC50值分别为142.67和40.62 μmol·L-1。Wang等[50, 51]合成了一系列caudatin衍生物, SAR研究表明3-羟基是活性所必需的基团, 但也产生了细胞毒性。将肉桂酸连接到65上得到化合物66, 其抑制HBsAg分泌、HBeAg分泌和HBV DNA复制的EC50值分别为5.52、5.52和2.44 μmol·L-1。初步机制认为66是通过干扰HBV启动子和增强子来影响HBV转录而发挥抗病毒作用。
|
HCV是由包膜为球形的RNA病毒, 核心是一条RNA单链, 由核衣壳包被。围绕衣壳的是糖蛋白囊膜, 表面蛋白嵌插其中。其生命周期主要分为吸附与融合、翻译和RNA复制、组装、出芽和释放等。
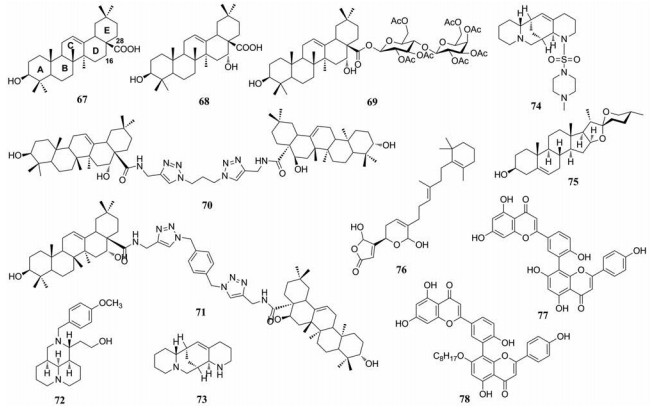
3.1 萜类化合物周德敏课题组及合作者一直致力于开发三萜类HCV侵入抑制剂。齐墩果酸(OA, 67)能较弱地抑制HCV进入宿主细胞, EC50值为10 μmol·L-1。SAR研究发现, C-16的羟基化(EA, 68)增强了抑制作用, 在28-羧基上连接二糖后(69) EC50值提高至0.3 μmol·L-1, 而且还除去了溶血不良反应。通过三唑的连接体形成的三萜二聚体(70、71)也显著提高了抑制作用, EC50约为10 nmol·L-1。机制研究表明, 这类三萜化合物通过与HCV包膜蛋白E2结合, 中断E2与其受体CD81之间的相互作用(图 3), 从而阻断病毒和宿主细胞的识别[52-54]。

|
Figure 3 The mechanism of triterpenes |
宋丹青和邓洪斌团队对三环苦参碱类化合物库进行筛选, 发现具有新结构骨架的HCV抑制剂, 代表化合物为N-苄基苦参碱(EC50 = 22.7 ± 0.67 μmol·L-1)[44]。然后以其为先导化合物对C-11位侧链的长度进行了研究, 发现该位的丁基链缩短为乙基链后, 活性未有显著变化, 化合物72显示出较好的抗HCV活性, EC50值为3.2 μmol·L-1, SI值为96.6[55]。72通过使热应激同源物70 (Hsc70) mRNA失去稳定性来下调转录后宿主Hsc70表达的水平, 从而阻断HCV复制[43]。
另外, 该团队还发现从苦豆子中提取的喹嗪类生物碱aloperine (73)具有良好的抗HCV活性, EC50值为4.32 μmol·L-1, SI值为54.5。进一步的结构改造得到衍生物74对野生株和突变株达到微摩尔的抑制活性(EC50 = 3.56±1.34 μmol·L-1), 并且可与直接抗病毒药物协同作用抑制HCV复制, 其主要机制可能靶向宿主组分。此外, 它还具有良好的口服药代动力学和安全性, 具有高度成药性[56]。
3.3 其他类化合物薯蓣皂苷元(75)是生产甾体激素类药物的重要化学原料。Wang等[57]发现75在低微摩尔浓度下抑制HCV复制, EC50值为3.8 μmol·L-1, 并未观察到细胞毒性。通过定量实时逆转录酶PCR和Western印迹分析, 发现它显著降低了病毒RNA和病毒蛋白的水平。此外, 它还减少了信号转导和转录激活因子的磷酸化, 与干扰素-R的组合后可以产生累加效应。
HCV的NS3蛋白因为具有核苷三磷酸酶(NTPase)和RNA解旋酶两种活性, 是抗HCV药的理想靶标。Salam等[58]利用高通量筛选光诱导电子转移(PET)系统, 从海绵提取物中得到了RNA解旋酶抑制剂manoalide (76)。深入研究76发现它以剂量依赖性方式抑制NS3蛋白的RNA解旋酶和ATP酶活性, 并且不影响NS3 ATP酶活性的表观Km值, 表现为非竞争性抑制。
Ma等[59]利用分子建模与计算机筛选确定银杏叶中的双黄酮amentoflavone 1a (77)为JAK2的Ⅱ型抑制剂, 可抑制HCV RNA的产生, EC50值大于50 μmol·L-1。随后他们通过计算机辅助优化策略, 在77的结构中引入一个或多个脂肪侧链使其更充分地占据疏水口袋, 辛基取代的衍生物78与先导化合物相比, 抗HCV活性明显增强(EC50 = 3.1 ± 0.8 μmol·L-1)。
4 抗流感病毒(influenza virus)流感病毒属于RNA病毒, 主要由核心(内含遗传物质及其复制所需的各种酶)、基质蛋白和包膜组成。病毒感染宿主细胞后, 经过吸附、内吞、融合、复制、翻译、装配、出芽、释放等过程复制病毒。目前小分子抗流感病毒药物主要包括M2离子通道阻滞剂、神经氨酸酶(NA)抑制剂、聚合酶PA亚基抑制剂、PB2亚基抑制剂以及一些广谱抗病毒药物。
|
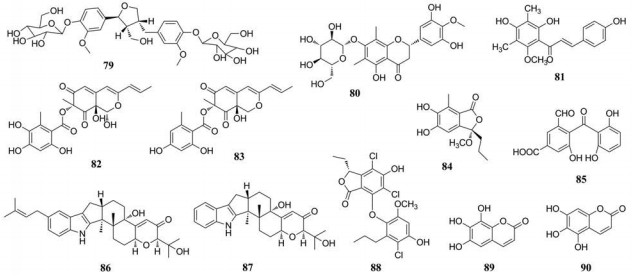
板蓝根作为中国传统药草, 已被广泛应用于治疗流感、流行性肝炎和乙型脑炎等。研究证明落叶松脂醇是板蓝根抗病毒的活性物质, Yang等[60]确证了落叶松脂醇衍生物clemastanin B (79), 在体外可显著抑制多种亚型的流感病毒, 其中, 抑制猪源流感病毒H1N1和禽流感病毒H7N3的IC50值分别为0.087和0.088 mg·mL-1。
Li等[61]从Matteuccia struthiopteris (L.) Todar的根茎中分离出黄酮类化合物80对H1N1流感病毒的神经氨酸酶具有显著的抑制活性, IC50值为6.8 ± 1.1 μmol·L-1 (阳性对照利巴韦林的IC50值为19.7 ± 1.0 μmol·L-1), SI值为34.4。通过分子对接研究80与神经氨酸酶的结合构象, 发现它可以与6个残基形成6个氢键, 能够与神经氨酸酶紧密结合。
Dao等[62]从Cleistocalyx operculatus的芽的甲醇提取物中分离出多种C-甲基化黄酮类化合物, 通过神经氨酸酶抑制实验发现查尔酮骨架的化合物81对甲型H1N1和奥司他韦抗性突变株(H274Y)的神经氨酸酶的抑制活性最强, IC50值分别为8.15 ± 1.05和3.31 ± 1.34 μmol·L-1, 动力学研究发现81为非竞争性抑制剂。
在强酸性条件下存活的真菌的次级代谢产物具有多种生物活性, 朱伟明课题组从中国云南红壤中分离出耐酸真菌Penicillium purpurogenum JS03-21, 发现它在pH 2的条件下发酵产生的次级代谢物要比在pH 7条件下的次级代谢产物具有更强的抗H1N1活性, 研究人员从前者的提取液中分离得到具有抗H1N1活性的化合物82~85, EC50值在58.6~85.3 μmol·L-1之间[63]。该课题组又从红树林土壤分离出耐酸性真菌Penicillium camemberti OUCMDZ-1492, 在其发酵液中分离得到吲哚-二萜类化合物86、87, 它们表现出更强的抗H1N1的活性, EC50值分别为6.6和17.7 μmol·L-1。SAR研究表明, 3-氧代、4b-羟基和9-异戊烯基取代会增加化合物的抗病毒活性[64]。
Niu等[65]从深海真菌Spiromastix sp中分离出一系列新型酚内酯, 大多数化合物对WSN流感病毒具有抑制活性, 且细胞毒性较低, 化合物88对A型和B型流感病毒的抗病毒活性最强, EC50值为6.0 ± 0.2 μmol·L-1。研究发现其作用机制可能是化合物与血凝素蛋白(HA)结合, 并破坏HA-唾液酸受体相互作用, 阻断病毒的附着和侵入。
Botta等[66]报道了儿茶酚衍生物对DNA和RNA病毒具有抑制活性, 发现生育酚氢醌衍生物是通过调节细胞的内部氧化还原电位抑制病毒复制[67]。为评价香豆素氧化物在抗病毒中的作用, 他们合成了儿茶酚和邻苯三酚衍生物, 通过血细胞凝集试验测定抗病毒活性, 发现具有邻苯三酚结构的香豆素衍生物(89、90)的抗病毒活性最强, EC50值分别为8.7和2.5 μmol·L-1[68]。
Mair等[69]在体外细胞病变效应抑制试验(CPE)中, 发现Burkea africana的树皮提取物具有抗流感病毒活性, EC50值为5.5 μg·mL-1, 随后分离出多种新的三萜皂苷, 齐墩果烷型三萜皂苷91 (线性三糖)和92 (支链四糖)的抗流感病毒活性较强, EC50值在0.05~0.27 μmol·L-1之间。
Wang等[70]从海洋真菌Cochliobolus lunatus SCSIO41401中分离出一种新的螺环γ-内酰胺93, 其对多种流感病毒株均有明显的抑制活性, EC50值在1.2~5.5 μmol·L-1之间。研究人员通过均相时间分辨荧光技术、表面等离子共振技术以及分子对接确定了93是通过结合聚合酶PB2蛋白的“帽”结构的高度保守区域来抑制蛋白活性, 从而抑制甲型流感病毒的复制。
|
尽管疱疹病毒家族的病毒之间存在形态和宿主的差异, 但核心部分都是双链DNA; 核心外包裹有衣壳蛋白, 外层还有糖蛋白镶嵌的脂质双层。疱疹病毒家族主要分为单纯疱疹病毒(HSV)、水痘-带状疱疹病毒(VZV)、人类疱疹病毒第四型(EBV)、巨细胞病毒(HCMV)等。
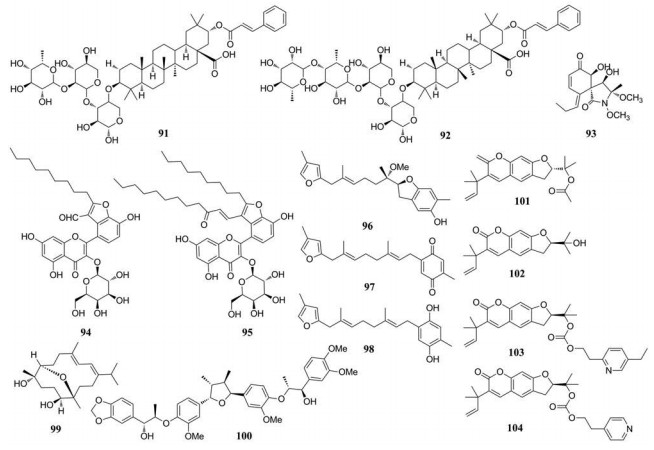
高昊课题组及合作者前期从鱼腥草中分离得到6种新型黄酮类化合物, 化合物houttuynoid A (94)、houttuynoid M (95)表现出有效的抗单疱疹病毒(HSV)活性, EC50值分别为12.42和17.72 μmol·L-1, 阳性对照药阿昔洛韦的EC50值为0.15 μmol·L-1 [71, 72]。机制研究发现, 94是通过阻断病毒膜融合来抑制HSV-1感染[73, 74]。
Cheng等[75]从软珊瑚S. capillosa的代谢产物中分离出多种呋喃糖萜类以及二萜类化合物, 通过体外的抗人巨细胞病毒测定发现呋喃糖萜类96和二萜化合物97、98表现出较强的抗病毒活性。他们还从海洋生物Octocoral Sarcophyton ehrenbergi中分离出西松烷化合物99, 在体外实验中表现出抗HCMV的活性, EC50值为4.7 μmol·L-1 [76]。
Cui等[77]从草本植物Saururus chinensis中提取分离得到28种木脂素, 并使用EBV裂解复制测定法进行活性测定, 发现多种化合物对EBV病毒具有抑制活性。化合物100表现出最强的抑制活性, EC50值为1.72 μmol·L-1, 且细胞毒性低(CC50 > 200 μmol·L-1), SI值大于116.4, 这也是首次发现木脂素类化合物具有抑制EBV裂解复制的活性。
从芸香科植物Ruta graveolens L中分离出的香豆素(+)-rutamarin (101)具有抑制EBV裂解复制的作用, EC50值为7.0 μmol·L-1 [78]。Lin等[79]从Ruta graveolens中分离出101的衍生物(-)-chalepin (102), 其EC50值为69.9 μmol·L-1。随后他们以102为起始原料合成了28种(-)-rutamarin衍生物, 多个衍生物的抗EBV活性要比101更强。其中, 化合物103、104的EC50值分别为1.5和0.32 μmol·L-1, SI值分别为801和211。
Ray等[80]发现从水稻(Oryza sativa)分离出的硫酸化葡聚糖具有强抗病毒活性, 其EC50值在2.44~6.5 μmol·L-1之间。与一般的葡聚糖(EC50 = 12.48 ± 0.60 μmol·L-1)相比, 硫酸化葡聚糖的抗HCMV病毒活性得到增强, 对疱疹病毒科的其他成员具有中等活性, 但对不相关的病毒没有活性。
|
肠病毒是一群病毒的总称, 最为常见的是EV71型, 俗称手足口病, 好发于五岁以下的儿童。此文主要介绍抗EV71和抗柯萨奇病毒(CVB)的天然产物研究进展。
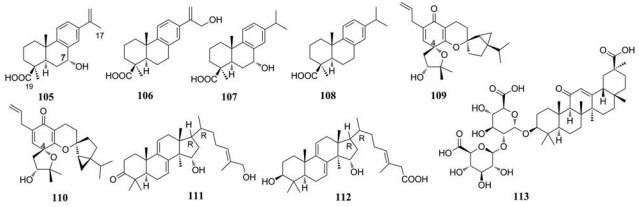
庾石山课题组从八角茴香的根中分离出多种倍半萜、二萜化合物, 并评价了化合物在体外的抗柯萨奇病毒(CVB)活性。二萜化合物105~108对多种柯萨奇病毒均表现出较好的抗病毒活性。SAR研究表明, 苯环上连有异丙烯基或乙酰基对活性或细胞毒性没有影响, 而在C-7或17位引入羟基或者在C-19位引入羧基可使抗病毒活性增加, 细胞毒性降低[81, 82]。
该课题组又分离得到具有二氧嘧啶骨架的螺旋藻酮化合物109和110, 它们对CVB3均有明显的抑制活性, EC50值分别为11.11和3.70 μmol·L-1[83]。深入研究其构型对生物活性的影响, 发现当C-4位的构型为R时, 大多数衍生物对CVB表现出有效的抑制活性, EC50值在1.88~4.29 μmol·L-1之间[84]。
Zhang等[85]发现两种灵芝三萜类化合物GLTA (111)、GLTB (112)在人横纹肌肉瘤(RD)细胞中表现出较强的抗EV71病毒活性, 但无细胞毒性。研究人员发现111和112通过与病毒颗粒相互作用来阻止病毒对细胞的吸附, 还通过阻断EV71脱壳抑制病毒RNA复制从而阻止病毒感染。
Chen等[86]发现荆芥(Schizonepeta tenuifolia Briq) (STE)的水提取物具有抗EV71病毒活性。机制研究发现, STE可以干扰病毒与宿主细胞的附着; 抑制真核翻译起始因子(eIF4G)的切割, 并阻碍异质型核糖核蛋白A1 (hnRNP A1)的细胞质转位。还可以清除活性氧(ROS)和抑制p38激酶磷酸化, 具有强大的抗EV71活性。
Kuo等[87]报道了甘草(Glycyrrhiza uralensis Fisch)的水提取物对EV71感染具有很强的抗病毒活性, EC50值为0.056 mg·mL-1。Wang等[88]证明了甘草酸(GA, 113)是甘草水提取物中的抗病毒成分, 但其抗病毒机制仍不清楚。
7 抗基孔肯雅病毒(CHIKV)基孔肯雅病毒(CHIKV)是一种主要由伊蚊传播的球形包膜单股正链RNA病毒, 含有多种非结构蛋白和结构蛋白。CHIKV感染可引起基孔肯雅热(Chikungunya fever), 主要表现为突发高热(39 ℃~40 ℃)、斑丘疹和持续性关节疼痛, 并伴有高病毒血症和抗原血症。
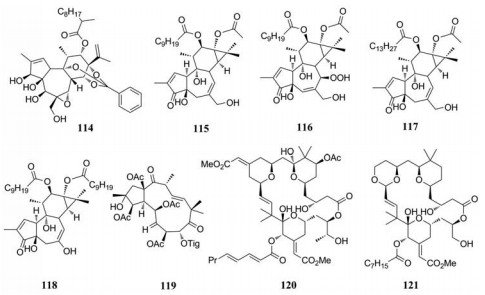
Marc Litaudon课题组通过生物测定所指导的纯化方法研究了多种大戟科植物, 得到了多种CHIKV抑制剂。从Trigonostemon cherrieri的树皮中分离出高度氧化的daphnane型二萜原酸酯(DDO, 114)表现出最强的抗CHIKV活性(EC50 = 0.6 ± 0.1 μmol·L-1, SI = 71.7)[89, 90]。从Croton mauritianus Lam的叶子的提取物中分离得到了tigliane二萜类化合物115、116, EC50值分别为2.4 ± 0.3和4.0 ± 0.8 μmol·L-1, 机制研究发现它们是通过抑制HIV颗粒进入T4淋巴细胞而表现出强烈的抗HIV活性[91]。与此同时他们也对多种类似于tigliane型二萜的化合物进行了生物活性测定, 化合物117被证明是一种非常有效的抑制剂, EC50值约为3 nmol·L-1, SI值接近2 000[92]。
该课题组更进一步地研究二萜类化合物, 评估了一系列市售天然二萜类化合物的抗病毒活性。其中, phorbol-12, 13-didecanoate (118)最有效, EC50值为6.0 ± 0.9 nmol·L-1, SI值为686, 其靶标可能是蛋白激酶C (PKC)[4]。
该课题组还从Euphorbia amygdaloides的乙酸乙酯提取物中分离得到多种jatrophane型酯类, 化合物119被证明是CHIKV、HIV-1和HIV-2病毒复制的高效选择性抑制剂(EC50值为0.76 μmol·L-1, IC50值分别为0.34和0.043 μmol·L-1)。初步的SAR表明, 化合物的抑制作用和选择性对jatrophane骨架上的取代基非常敏感, 可能涉及PKC依赖性机制[93]。
苔藓抑素1 (120)本身是一种有效的泛蛋白激酶C (PKC)调节剂, Staveness等[94, 95]报道120对CHIKV病毒无抑制活性, 这说明之前此类化合物的抗CHIKV活性不仅仅是由PKC调节介导的, 可能有尚未确定的靶点。但其水杨酸盐衍生的苔藓抑素类似物(121)却具有比母体化合物更强的活性, 也为PKC非依赖性途径的参与提供了证据。
|
|
登革病毒属于黄病毒科, 基因组全长约11 kb, 为单链正链RNA病毒。病毒颗粒由内而外分为膜蛋白M、包膜、包膜蛋白E、脂蛋白包膜。DENV的发病机制尚不明确, 迄今尚无特效药物。
Bourjot等[96]对Flacourtia ramontchi的乙酸乙酯提取物使用生物测定的分离方法获得6种新的酚类糖苷以及桦木酸3β-咖啡酸(122)。基于DENV NS5聚合酶的测试, 通过监测标记的放射性鸟苷掺入聚胞嘧啶RNA模板来测定聚合酶活性, 大部分酚类糖苷并不显示活性, 而化合物122表现出显著的活性, IC50值为0.85 ± 0.1 μmol·L-1。
从鳄梨(Persea americana)果实中提取的天然产物(2R, 4R)-1, 2, 4-三羟基十七烷-16-炔(THHY, 123)能够以浓度依赖性的方式抑制DENV-2复制。Wu等[97]已经证实123是通过上调NF-κB介导的IFN反应来抑制DENV复制, 并且它还能增加感染DENV的哺乳小鼠存活率, 使鳄梨果实成为治疗DENV感染和DENV相关疾病的潜在膳食资源。
Allard等[98]从苜蓿科植物Cryptocarya chartacea的树皮中分离出多种次生代谢物, 多个化合物在DENV聚合酶测定中表现出明显的抗病毒活性, EC50在1.8 ± 1.2~4.2 ± 0.1 μmol·L-1之间, 而且它们在10 μg·mL-1的KB细胞系中没有细胞毒性。其中, chartaceone D (124)的抗病毒活性最强, 此类结构代表了一类新的DENV聚合酶的非核苷类抑制剂。
Estoppey等[99]在真菌中发现了cavinafungin (125), 它靶向内质网定位的信号肽酶, 可切割宿主和病毒蛋白的信号基序。125能够对抗寨卡病毒和所有4种登革热病毒血清型, 但对其他类型的病毒却是无效。SAR研究表明, 125的醛结构的丧失将导致抗病毒活性降低和治疗指数急剧下滑。
9 抗其他病毒呼吸道合胞病毒(RSV)是婴儿和儿童中严重的病毒性下呼吸道疾病的最常见的病原体。王长云课题组及合作者从柳珊瑚上的真菌Aspergillus sp.培养液中分离出异戊二烯二氢喹诺酮的衍生物126表现出较强的抗RSV活性, EC50值为42 nmol·L-1, 比阳性对照利巴韦林强(EC50 = 20 μmol·L-1)强约500倍, 并且显示出较高的选择性指数(SI = 520)[100]。又从柳珊瑚Echinogorgia rebekka的乙酸乙酯提取物中分离得到一对差向异构体(127、128), 显示出相等的抗RSV活性, EC50值为0.19 μmol·L-1。有趣的是, C-25的构型可能与毒性有某种关系, 所以它们对Hep-2细胞表现出不同的毒性, CC50值分别为0.38和24.4 μmol·L-1, 因此, 128表现出更高的治疗指数[101]。

|
埃博拉病毒是一种能引起人类和灵长类动物产生埃博拉出血热的烈性传染病毒, 有很高的死亡率。Zhang等[102]以含有内环骨架的苦豆碱作为先导化合物, 合成了23种新的苦豆碱衍生物, 并使用假型病毒模型评估了它们的抗病毒活性, 包括埃博拉病毒(EBOV)和马尔堡病毒(MARV)。化合物129在体外和体内均表现出最有效的抗EBOV和抗MARV活性(EC50值分别为4.8和7.1 μmol·L-1), 被认为具有广谱抗丝状病毒活性。机制研究表明, 129主要是通过抑制宿主细胞的半胱氨酸组织蛋白酶B的活性来阻止病毒进入细胞。
人鼻病毒(HRV)是导致轻度上呼吸道疾病的主要病原体, 被认为是人类中最常见的感染因子。Fois等[103]发现柴胡叶子的二氯甲烷提取物抑制人鼻病毒(HRV)血清型14和39的复制, 分离纯化提取物得到化合物130和131, 两者表现出对HRV病毒复制的选择性抑制, EC50值分别为1.8 ± 0.02和2.4 ± 0.04 μmol·L-1。机制研究表明, 该类化合物不仅是衣壳结合剂, 干扰病毒复制的早期阶段, 而且也是晚期复制抑制剂。分子模拟研究也证明了这些化合物能够结合到HRV VP1蛋白的疏水口袋中。
天花(痘病毒)被视为大规模杀伤性武器的几种病毒之一, 目前虽然通过免疫接种等已基本消灭, 但出于新型防御战略的需要, 仍须研制出抗痘病毒的药物。Pirrung等[104]通过对缩胺基硫脲类化合物库进行高通量筛选得到化合物132, EC50值为6.0 ± 2.9 μmol·L-1, CC50值为181 ± 67 μmol·L-1, 研究人员猜测该类化合物通过增强病毒复制后转录的伸长, 导致形成比正常转录更长的转录本。
10 总结与展望综上所示, 天然产物是发现抗病毒药物先导化合物与候选药物的重要源泉。尽管如此, 不容否认的是, 目前天然产物的研究及发展仍面临着瓶颈, 首先是难以应用筛选技术得到先导化合物, 其次是复杂天然产物进行全合成较为困难, 最重要的是大多数有活性的天然产物局限于体外细胞实验, 鲜有动物体内及临床试验。通过利用天然产物化合物库以及计算机虚拟筛选等技术, 快速得到活性良好的先导化合物; 通过将人工智能技术与天然产物全合成相融合探索复杂产物的合成路线; 最后要推进高活性低毒性天然产物的活性评价, 进行动物实验。

|
需要指出的是, 人体在感染病毒后, 体内形成病毒潜伏库, 现有的绝大多数抗病毒药物不能完全清除病毒。为了根治病毒性传染病, 潜伏激活(shock and kill)是彻底清除病毒的重要途径, 也是极具潜力的抗病毒前沿方向。在此文中, 笔者注意到许多天然产物在体外细胞模型中表现出较强的潜伏激活活性, 相信随着研究的深入, 天然产物将在病毒潜伏激活剂研究领域大放异彩。
中药在疾病治疗中具有独特的优势, 但也面临着发掘不够、传承不足等问题。2019年10月26日, 国务院发布了《中共中央国务院关于促进中医药传承创新发展的意见》, 在国家大力支持开发特色原创药物、大力弘扬中药的有利背景下, 需要结合现代药理学、药效学、分子生物学、化学生物学等新技术, 应用高通量筛选、虚拟筛选等新理论, 对我国特有天然产物及具有“清瘟解毒”等疗效的传统中药进行多角度、多层面组合研究, 早日研制出新型低毒高效的抗病毒药物。
| [1] |
Siegel L, Gulick RM. New antiretroviral agents[J]. Curr Infect Dis Rep, 2007, 9: 243-251. DOI:10.1007/s11908-007-0038-8 |
| [2] |
Fang PL, Cao YL, Yan H, et al. Lindenane disesquiterpenoids with anti-HIV-1 activity from Chloranthus japonicus[J]. J Nat Prod, 2011, 74: 1408-1413. |
| [3] |
Yang Y, Cao YL, Liu HY, et al. Shizukaol F: a new structural type inhibitor of HIV-1 reverse transcriptase RNase H[J]. Acta Pharm Sin (药学学报), 2012, 47: 1011-1016. |
| [4] |
Nothias-Scaglia LF, Pannecouque C, Renucci F, et al. Antiviral activity of diterpene of esters on Chikungunya virus and HIV replication[J]. J Nat Prod, 2015, 78: 1277-1283. DOI:10.1021/acs.jnatprod.5b00073 |
| [5] |
Pardo-Vargas A, Ramos FA, Cirne-Santos CC, et al. Semi-synthesis of oxygenated dolabellane diterpenes with highly in vitro anti-HIV-1 activity[J]. Bioorg Med Chem Lett, 2014, 24: 4381-4383. DOI:10.1016/j.bmcl.2014.08.019 |
| [6] |
Huang YS, Lu Y, Chen CH, et al. Potent anti-HIV ingenane diterpenoids from Euphorbia ebracteolata[J]. J Nat Prod, 2019, 82: 1587-1592. DOI:10.1021/acs.jnatprod.9b00088 |
| [7] |
Liu Q, Li W, Huang L, et al. Identification, structural modification, and dichotomous effects on human immunodeficiency virus type 1 (HIV-1) replication of ingenane esters from Euphorbia kansui[J]. Eur J Med Chem, 2018, 156: 618-627. DOI:10.1016/j.ejmech.2018.07.020 |
| [8] |
Jiang G, Mendes EA, Kaiser P, et al. Synergistic reactivation of latent HIV expression by ingenol-3-angelate, PEP005, targeted NF-κB signaling in combination with JQ1 induced p-TEFb activation[J]. PLoS Pathog, 2015, 11: e1005066. DOI:10.1371/journal.ppat.1005066 |
| [9] |
Pandeló José D, Bartholomeeusen K, da Cunha RD, et al. Reactivation of latent HIV-1 by new semi-synthetic ingenol esters[J]. Virology, 2014, 462-463: 328-339. DOI:10.1016/j.virol.2014.05.033 |
| [10] |
Huang L, Ho P, Yu J, et al. Picomolar dichotomous activity of gnidimacrin against HIV-1[J]. PLoS One, 2011, 6: e26677. DOI:10.1371/journal.pone.0026677 |
| [11] |
Vidal V, Potterat O, Louvel S, et al. Library-based discovery and characterization of daphnane diterpenes as potent and selective HIV inhibitors in Daphne gnidium[J]. J Nat Prod, 2012, 75: 414-419. DOI:10.1021/np200855d |
| [12] |
Zhang D, Guo J, Zhang M, et al. Oxazole-containing diterpenoids from cell cultures of Salvia miltiorrhiza and their anti-HIV1 activities[J]. J Nat Prod, 2017, 80: 3241-3246. DOI:10.1021/acs.jnatprod.7b00659 |
| [13] |
Osorio AA, Muñóz A, Torres-Romero D, et al. Olean-18-ene triterpenoids from Celastraceae species inhibit HIV replication targeting NF-κB and Sp1 dependent transcription[J]. Eur J Med Chem, 2012, 52: 295-303. |
| [14] |
Callies O, Bedoya LM, Beltrán M, et al. Isolation, structural modification, and HIV inhibition of pentacyclic lupane-type triterpenoids from Cassine xylocarpa and Maytenus cuzcoina[J]. J Nat Prod, 2015, 78: 1045-1055. DOI:10.1021/np501025r |
| [15] |
Zhang SY, Meng L, Gao WY, et al. Advances on biological activities of coumarins[J]. China J Chin Mater Med (中国中药杂志), 2005, 30: 410-414. |
| [16] |
Esposito F, Ambrosio FA, Maleddu R, et al. Chromenone derivatives as a versatile scaffold with dual mode of inhibition of HIV-1 reverse transcriptase-associated Ribonuclease H function and integrase activity[J]. Eur J Med Chem, 2019, 182: 111617. DOI:10.1016/j.ejmech.2019.111617 |
| [17] |
Sonar VP, Corona A, Distinto S, et al. Natural product-inspired esters and amides of ferulic and caffeic acid as dual inhibitors of HIV-1 reverse transcriptase[J]. Eur J Med Chem, 2017, 130: 248-260. DOI:10.1016/j.ejmech.2017.02.054 |
| [18] |
Kashman Y, Gustafson KR, Fuller RW, et al. The calanolides, a novel HIV-inhibitory class of coumarin derivatives from the tropical rainforest tree, Calophyllum lanigerum[J]. J Med Chem, 1992, 35: 2735-2743. DOI:10.1021/jm00093a004 |
| [19] |
Xue H, Lu X, Zheng P, et al. Highly suppressing wild-type HIV-1 and Y181C mutant HIV-1 strains by 10-chloromethyl-11-demethyl-12-oxo-calanolide A with druggable profile[J]. J Med Chem, 2010, 53: 1397-1401. DOI:10.1021/jm901653e |
| [20] |
Dong B, Ma T, Zhang T, et al. Anti-HIV-1 activity and structure-activity relationship of pyranocoumarin analogs[J]. Acta Pharm Sin (药学学报), 2011, 46: 35-38. |
| [21] |
Zhang HJ, Rumschlag-Booms E, Guan YF, et al. Potent inhibitor of drug-resistant HIV1 strains identified from the medicinal plant Justicia gendarussa[J]. J Nat Prod, 2017, 80: 1798-1807. DOI:10.1021/acs.jnatprod.7b00004 |
| [22] |
Zhang HJ, Rumschlag-Booms E, Guan YF, et al. Anti-HIV diphyllin glycosides from Justicia gendarussa[J]. Phytochemistry, 2017, 136: 94-100. DOI:10.1016/j.phytochem.2017.01.005 |
| [23] |
Prasad S, Tyagi AK. Curcumin and its analogues: a potential natural compound against HIV infection and AIDS[J]. Food Funct, 2015, 6: 3412-3419. DOI:10.1039/C5FO00485C |
| [24] |
Zhang HS, Zhou Y, Wu MR, et al. Resveratrol inhibited Tat-induced HIV-1 LTR transactivation via NAD+-dependent SIRT1 activity[J]. Life Sci, 2009, 85: 484-489. DOI:10.1016/j.lfs.2009.07.014 |
| [25] |
Pal Singh I, Bharate SB. Phloroglucinol compounds of natural origin[J]. Nat Prod Rep, 2006, 23: 558-591. DOI:10.1039/b600518g |
| [26] |
Chauthe SK, Bharate SB, Sabde S, et al. Biomimetic synthesis and anti-HIV activity of dimeric phloroglucinols[J]. Bioorg Med Chem, 2010, 18: 2029-2036. |
| [27] |
Kamng'ona A, Moore JP, Lindsey G, et al. Inhibition of HIV-1 and M-MLV reverse transcriptases by a major polyphenol (3, 4, 5 tri-O-galloylquinic acid) present in the leaves of the South African resurrection plant, Myrothamnus flabellifolia[J]. J Enzyme Inhib Med Chem, 2011, 26: 843-853. DOI:10.3109/14756366.2011.566220 |
| [28] |
Chaves Valadão AL, Abreu CM, Dias JZ, et al. Natural plant alkaloid (emetine) inhibits HIV-1 replication by interfering with reverse transcriptase activity[J]. Molecules, 2015, 20: 11474-11489. DOI:10.3390/molecules200611474 |
| [29] |
McCormick JL, McKee TC, Cardellina JH, et al. HIV inhibitory natural products. 26. Quinoline alkaloids from Euodia roxburghiana[J]. J Nat Prod, 1996, 59: 469-471. DOI:10.1021/np960250m |
| [30] |
Ahmed N, Brahmbhatt KG, Sabde S, et al. Synthesis and anti-HIV activity of alkylated quinoline 2, 4-diols[J]. Bioorg Med Chem, 2010, 18: 2872-2879. DOI:10.1016/j.bmc.2010.03.015 |
| [31] |
Jadulco RC, Pond CD, Van Wagoner RM, et al. 4-Quinolone alkaloids from Melochia odorata[J]. J Nat Prod, 2014, 77: 183-187. DOI:10.1021/np400847t |
| [32] |
Esposito F, Carli I, Del Vecchio C, et al. Sennoside A, derived from the traditional chinese medicine plant Rheum L., is a new dual HIV-1 inhibitor effective on HIV-1 replication[J]. Phytomedicine, 2016, 23: 1383-1391. DOI:10.1016/j.phymed.2016.08.001 |
| [33] |
He X, Wang Y, Luo RH, et al. Dimeric pyranonaphthoquinone glycosides with anti-HIV and cytotoxic activities from a soil-derived streptomyces[J]. J Nat Prod, 2019, 82: 1813-1819. DOI:10.1021/acs.jnatprod.9b00022 |
| [34] |
Lee J, Oh WK, Ahn JS, et al. Prenylisoflavonoids from Erythrina senegalensis as novel HIV-1 protease inhibitors[J]. Planta Med, 2009, 75: 268-270. |
| [35] |
Omolo JJ, Maharaj V, Naidoo D, et al. Bioassay-guided investigation of the Tanzanian plant Pyrenacantha kaurabassana for potential anti-HIV-active compounds[J]. J Nat Prod, 2012, 75: 1712-1716. |
| [36] |
Guo RH, Geng CA, Huang XY, et al. Synthesis of hemslecin A derivatives: a new class of hepatitis B virus inhibitors[J]. Bioorg Med Chem Lett, 2013, 23: 1201-1205. |
| [37] |
Ma WH, Huang H, Zhou P, et al. Schisanwilsonenes A-C, anti-HBV carotane sesquiterpenoids from the fruits of Schisandra wilsoniana[J]. J Nat Prod, 2009, 72: 676-678. DOI:10.1021/np8007864 |
| [38] |
Zhang Q, Jiang ZY, Luo J, et al. Anti-HBV agents. Part 2: Synthesis and in vitro anti-hepatitis B virus activities of alisol A derivatives[J]. Bioorg Med Chem Lett, 2009, 19: 2148-214=53. DOI:10.1016/j.bmcl.2009.02.122 |
| [39] |
Zhang Q, Jiang ZY, Luo J, et al. Anti-HBV agents. Part 3: Preliminary structure-activity relationships of tetra-acylalisol A derivatives as potent hepatitis B virus inhibitors[J]. Bioorg Med Chem Lett, 2009, 19: 6659-6665. DOI:10.1016/j.bmcl.2009.10.006 |
| [40] |
Zhang YB, Luo D, Yang L, et al. Matrine-type alkaloids from the roots of Sophora flavescens and their antiviral activities against the hepatitis B virus[J]. J Nat Prod, 2018, 81: 2259-2265. DOI:10.1021/acs.jnatprod.8b00576 |
| [41] |
Zhang YB, Zhang XL, Chen NH, et al. Four matrine-based alkaloids with antiviral activities against HBV from the seeds of Sophora alopecuroides[J]. Org Lett, 2017, 19: 424-427. DOI:10.1021/acs.orglett.6b03685 |
| [42] |
Zhang YB, Yang L, Luo D, et al. Sophalines E-I, five quinolizidine-based alkaloids with antiviral activities against the hepatitis B virus from the seeds of Sophora alopecuroides[J]. Org Lett, 2018, 20: 5942-5946. DOI:10.1021/acs.orglett.8b02637 |
| [43] |
Peng ZG, Fan B, Du NN, et al. Small molecular compounds that inhibit hepatitis C virus replication through destabilizing heat shock cognate 70 messenger RNA[J]. Hepatology, 2010, 52: 845-853. DOI:10.1002/hep.23766 |
| [44] |
Du NN, Li X, Wang YP, et al. Synthesis, structure-activity relationship and biological evaluation of novel N-substituted matrinic acid derivatives as host heat-stress cognate 70 (Hsc70) down-regulators[J]. Bioorg Med Chem Lett, 2011, 21: 4732-4735. DOI:10.1016/j.bmcl.2011.06.071 |
| [45] |
Wan CX, Zhang PH, Luo JG, et al. Homoflavonoid glucosides from Ophioglossum pedunculosum and their anti-HBV activity[J]. J Nat Prod, 2011, 74: 683-689. DOI:10.1021/np100745z |
| [46] |
Ying C, Li Y, Leung CH, et al. Unique antiviral mechanism discovered in anti-hepatitis B virus research with a natural product analogue[J]. Proc Natl Acad Sci U S A, 2007, 104: 8526-8531. |
| [47] |
Yang L, Shi LP, Chen HJ, et al. Isothiafludine, a novel non-nucleoside compound, inhibits hepatitis B virus replication through blocking pregenomic RNA encapsidation[J]. Acta Pharmacol Sin, 2014, 35: 410-418. DOI:10.1038/aps.2013.175 |
| [48] |
Janmanchi D, Tseng YP, Wang KC, et al. Synthesis and the biological evaluation of arylnaphthalene lignans as anti-hepatitis B virus agents[J]. Bioorg Med Chem, 2010, 18: 1213-1226. DOI:10.1016/j.bmc.2009.12.038 |
| [49] |
Zhang F, Wang G. A review of non-nucleoside anti-hepatitis B virus agents[J]. Eur J Med Chem, 2014, 75: 267-281. DOI:10.1016/j.ejmech.2014.01.046 |
| [50] |
Wang LJ, Geng CA, Ma YB, et al. Synthesis, structure-activity relationships and biological evaluation of caudatin derivatives as novel anti-hepatitis B virus agents[J]. Bioorg Med Chem, 2012, 20: 2877-2888. DOI:10.1016/j.bmc.2012.03.023 |
| [51] |
Wang LJ, Geng CA, Ma YB, et al. Design, synthesis, and molecular hybrids of caudatin and cinnamic acids as novel anti-hepatitis B virus agents[J]. Eur J Med Chem, 2012, 54: 352-365. DOI:10.1016/j.ejmech.2012.05.012 |
| [52] |
Yu F, Wang Q, Zhang Z, et al. Development of oleanane-type triterpenes as a new class of HCV Entry Inhibitors[J]. J Med Chem, 2013, 56: 4300-4319. DOI:10.1021/jm301910a |
| [53] |
Wang H, Wang Q, Xiao SL, et al. Elucidation of the pharmacophore of echinocystic acid, a new lead for blocking HCV entry[J]. Eur J Med Chem, 2013, 64: 160-168. |
| [54] |
Yu F, Peng Y, Wang Q, et al. Development of bivalent oleanane-type triterpenes as potent HCV entry inhibitors[J]. Eur J Med Chem, 2014, 77: 258-268. DOI:10.1016/j.ejmech.2014.03.017 |
| [55] |
Li YH, Wu ZY, Tang S, et al. Evolution of matrinic ethanol derivatives as anti-HCV agents from matrine skeleton[J]. Bioorg Med Chem Lett, 2017, 27: 1962-1966. DOI:10.1016/j.bmcl.2017.03.025 |
| [56] |
Zhang X, Lv XQ, Tang S, et al. Discovery and evolution of aloperine derivatives as a new family of HCV inhibitors with novel mechanism[J]. Eur J Med Chem, 2018, 143: 1053-1065. DOI:10.1016/j.ejmech.2017.12.002 |
| [57] |
Wang YJ, Pan KL, Hsieh TC, et al. Diosgenin, a plant-derived sapogenin, exhibits antiviral activity in vitro against hepatitis C virus[J]. J Nat Prod, 2011, 74: 580-584. DOI:10.1021/np100578u |
| [58] |
Salam KA, Furuta A, Noda N, et al. Inhibition of hepatitis C virus NS3 helicase by manoalide[J]. J Nat Prod, 2012, 75: 650-654. DOI:10.1021/np200883s |
| [59] |
Ma DL, Chan DS, Wei G, et al. Virtual screening and optimization of Type Ⅱ inhibitors of JAK2 from a natural product library[J]. Chem Commun (Camb), 2014, 50: 13885-13888. DOI:10.1039/C4CC04498C |
| [60] |
Yang Z, Wang Y, Zheng Z, et al. Antiviral activity of Isatis indigotica root-derived clemastanin B against human and avian influenza A and B viruses in vitro[J]. Int J Mol Med, 2013, 31: 867-873. |
| [61] |
Li B, Ni Y, Zhu LJ, et al. Flavonoids from Matteuccia struthiopteris and their anti-influenza virus (H1N1) activity[J]. J Nat Prod, 2015, 78: 987-995. DOI:10.1021/np500879t |
| [62] |
Dao TT, Tung BT, Nguyen PH, et al. C-Methylated flavonoids from Cleistocalyx operculatus and their inhibitory effects on novel influenza A (H1N1) neuraminidase[J]. J Nat Prod, 2010, 73: 1636-1642. |
| [63] |
Wang H, Wang Y, Wang W, et al. Anti-influenza virus polyketides from the acid-tolerant fungus Penicillium purpurogenum JS03-21[J]. J Nat Prod, 2011, 74: 2014-2018. |
| [64] |
Fan Y, Wang Y, Liu P, et al. Indole-diterpenoids with anti-H1N1 activity from the aciduric fungus Penicillium camemberti OUCMDZ-1492[J]. J Nat Prod, 2013, 76: 1328-1336. DOI:10.1021/np400304q |
| [65] |
Niu S, Si L, Liu D, et al. Spiromastilactones: a new class of influenza virus inhibitors from deep-sea fungus[J]. Eur J Med Chem, 2016, 108: 229-244. DOI:10.1016/j.ejmech.2015.09.037 |
| [66] |
Botta G, Bizzarri BM, Garozzo A, et al. Carbon nanotubes supported tyrosinase in the synthesis of lipophilic hydroxytyrosol and dihydrocaffeoyl catechols with antiviral activity against DNA and RNA viruses[J]. Bioorg Med Chem, 2015, 23: 5345-5351. DOI:10.1016/j.bmc.2015.07.061 |
| [67] |
Saladino R, Barontini M, Crucianelli M, et al. Current advances in anti-influenza therapy[J]. Curr Med Chem, 2010, 17: 2101-2140. DOI:10.2174/092986710791299957 |
| [68] |
Bizzarri BM, Botta L, Capecchi E, et al. Regioselective IBX-mediated synthesis of coumarin derivatives with antioxidant and anti-influenza activities[J]. J Nat Prod, 2017, 80: 3247-3254. DOI:10.1021/acs.jnatprod.7b00665 |
| [69] |
Mair CE, Grienke U, Wilhelm A, et al. Anti-influenza triterpene saponins from the bark of Burkea africana[J]. J Nat Prod, 2018, 81: 515-523. DOI:10.1021/acs.jnatprod.7b00774 |
| [70] |
Wang J, Chen F, Liu Y, et al. Spirostaphylotrichin X from a marine-derived fungus as an antiinfluenza agent targeting RNA polymerase PB2[J]. J Nat Prod, 2018, 81: 2722-2730. DOI:10.1021/acs.jnatprod.8b00656 |
| [71] |
Chen SD, Gao H, Zhu QC, et al. Houttuynoids A-E, anti-herpes simplex virus active flavonoids with novel skeletons from Houttuynia cordata[J]. Org Lett, 2012, 14: 1772-1775. DOI:10.1021/ol300017m |
| [72] |
Li JJ, Chen GD, Fan HX, et al. Houttuynoid M, an anti-HSV active houttuynoid from Houttuynia cordata featuring a bis-houttuynin chain tethered to a flavonoid core[J]. J Nat Prod, 2017, 80: 3010-3013. DOI:10.1021/acs.jnatprod.7b00620 |
| [73] |
Jian J, Fan J, Yang H, et al. Total synthesis of the flavonoid natural product houttuynoid A[J]. J Nat Prod, 2018, 81: 371-377. DOI:10.1021/acs.jnatprod.7b00791 |
| [74] |
Li T, Liu L, Wu H, et al. Anti-herpes simplex virus type 1 activity of houttuynoid A, a new flavonoid from Houttuynia cordata Thunb[J]. Antiviral Res, 2017, 144: 273-280. DOI:10.1016/j.antiviral.2017.06.010 |
| [75] |
Cheng SY, Huang KJ, Wang SK, et al. Antiviral and anti-inflammatory metabolites from the soft coral Sinularia capillosa[J]. J Nat Prod, 2010, 73: 771-775. DOI:10.1021/np9008078 |
| [76] |
Cheng SY, Wang SK, Chiou SF, et al. Cembranoids from the Octocoral Sarcophyton ehrenbergi[J]. J Nat Prod, 2010, 73: 197-203. |
| [77] |
Cui H, Xu B, Wu T, et al. Potential antiviral lignans from the roots of Saururus chinensis with activity against Epstein-Barr virus lytic replication[J]. J Nat Prod, 2014, 77: 100-110. DOI:10.1021/np400757k |
| [78] |
Wu T, Wang Y, Yuan Y, et al. Antiviral activity of topoisomerase Ⅱ catalytic inhibitors against Epstein-Barr virus[J]. Antiviral Res, 2014, 107: 95-101. DOI:10.1016/j.antiviral.2014.05.003 |
| [79] |
Lin Y, Wang Q, Gu Q, et al. Semisynthesis of (-)-rutamarin derivatives and their inhibitory activity on Epstein-Barr virus lytic replication[J]. J Nat Prod, 2017, 80: 53-60. |
| [80] |
Ray B, Hutterer C, Bandyopadhyay SS, et al. Chemically engineered sulfated glucans from rice bran exert strong antiviral activity at the stage of viral entry[J]. J Nat Prod, 2013, 76: 2180-2188. DOI:10.1021/np4003977 |
| [81] |
Zhang GJ, Li YH, J DJ, et al. Anti-coxsackie virus B diterpenes from the roots of Illicium jiadifengpi[J]. Tetrahedron, 2013, 69: 1017-1023. |
| [82] |
Wang YD, Zhang GJ, Qu J, et al. Diterpenoids and sesquiterpenoids from the roots of Illicium majus[J]. J Nat Prod, 2013, 76: 1976-1983. DOI:10.1021/np400638r |
| [83] |
Ma SG, Gao RM, Li YH, et al. Antiviral spirooliganones A and B with unprecedented skeletons from the roots of Illicium oligandrum[J]. Org Lett, 2013, 15: 4450-4453. DOI:10.1021/ol401992s |
| [84] |
Zhao N, Ren X, Ren J, et al. Total syntheses of (-)-spirooliganones A and B and their diastereoisomers: absolute stereochemistry and inhibitory activity against coxsackie virus B3[J]. Org Lett, 2015, 17: 3118-3121. DOI:10.1021/acs.orglett.5b01419 |
| [85] |
Zhang W, Tao J, Yang X, et al. Antiviral effects of two Ganoderma lucidum triterpenoids against enterovirus 71 infection[J]. Biochem Biophys Res Commun, 2014, 449: 307-312. DOI:10.1016/j.bbrc.2014.05.019 |
| [86] |
Chen SG, Cheng ML, Chen KH, et al. Antiviral activities of Schizonepeta tenuifolia Briq. against enterovirus 71 in vitro and in vivo[J]. Sci Rep, 2017, 7: 935. DOI:10.1038/s41598-017-01110-x |
| [87] |
Kuo KK, Chang JS, Wang KC, et al. Water extract of Glycyrrhiza uralensis inhibited enterovirus 71 in a human foreskin fibroblast cell line[J]. Am J Chin Med, 2009, 37: 383-394. DOI:10.1142/S0192415X09006904 |
| [88] |
Wang J, Chen X, Wang W, et al. Glycyrrhizic acid as the antiviral component of Glycyrrhiza uralensis Fisch. against coxsackievirus A16 and enterovirus 71 of hand foot and mouth disease[J]. J Ethnopharmacol, 2013, 147: 114-121. DOI:10.1016/j.jep.2013.02.017 |
| [89] |
Allard PM, Leyssen P, Martin MT, et al. Antiviral chlorinated daphnane diterpenoid orthoesters from the bark and wood of Trigonostemon cherrieri[J]. Phytochemistry, 2012, 84: 160-168. |
| [90] |
Bourjot M, Leyssen P, Neyts J, et al. Trigocherrierin A, a potent inhibitor of Chikungunya virus replication[J]. Molecules, 2014, 19: 3617-3627. DOI:10.3390/molecules19033617 |
| [91] |
Corlay N, Delang L, Girard-Valenciennes E, et al. Tigliane diterpenes from Croton mauritianus as inhibitors of Chikungunya virus replication[J]. Fitoterapia, 2014, 97: 87-91. DOI:10.1016/j.fitote.2014.05.015 |
| [92] |
Bourjot M, Delang L, Nguyen VH, et al. Prostratin and 12-O-tetradecanoylphorbol 13-acetate are potent and selective inhibitors of Chikungunya virus replication[J]. J Nat Prod, 2012, 75: 2183-2187. DOI:10.1021/np300637t |
| [93] |
Nothias-Scaglia LF, Retailleau P, Paolini J, et al. Jatrophane diterpenes as inhibitors of Chikungunya virus replication: structure-activity relationship and discovery of a potent lead[J]. J Nat Prod, 2014, 77: 1505-1512. DOI:10.1021/np500271u |
| [94] |
Staveness D, Abdelnabi R, Schrier AJ, et al. Simplified bryostatin analogues protect cells from Chikungunya virus-induced cell death[J]. J Nat Prod, 2016, 79: 675-679. DOI:10.1021/acs.jnatprod.5b01016 |
| [95] |
Staveness D, Abdelnabi R, Near KE, et al. Inhibition of Chikungunya virus-induced cell death by salicylate-derived bryostatin analogues provides additional evidence for a PKC-independent pathway[J]. J Nat Prod, 2016, 79: 680-684. DOI:10.1021/acs.jnatprod.5b01017 |
| [96] |
Bourjot M, Leyssen P, Eydoux C, et al. Flacourtosides A-F, phenolic glycosides isolated from Flacourtia ramontchi[J]. J Nat Prod, 2012, 75: 752-758. DOI:10.1021/np300059n |
| [97] |
Wu YH, Tseng CK, Wu HC, et al. Avocado (Persea americana) fruit extract (2R, 4R)-1, 2, 4-trihydroxyheptadec-16-yne inhibits dengue virus replication via upregulation of NF-κB-dependent induction of antiviral interferon responses[J]. Sci Rep, 2019, 9: 423. DOI:10.1038/s41598-018-36714-4 |
| [98] |
Allard PM, Dau ET, Eydoux C, et al. Alkylated flavanones from the bark of Cryptocarya chartacea as Dengue virus NS5 polymerase inhibitors[J]. J Nat Prod, 2011, 74: 2446-2453. DOI:10.1021/np200715v |
| [99] |
Estoppey D, Lee CM, Janoschke M, et al. The natural product cavinafungin selectively interferes with Zika and Dengue virus replication by inhibition of the host signal peptidase[J]. Cell Rep, 2017, 19: 451-460. DOI:10.1016/j.celrep.2017.03.071 |
| [100] |
Chen M, Shao CL, Meng H, et al. Anti-respiratory syncytial virus prenylated dihydroquinolone derivatives from the gorgonian-derived fungus Aspergillus sp. XS-20090B15[J]. J Nat Prod, 2014, 77: 2720-2724. DOI:10.1021/np500650t |
| [101] |
Cao F, Shao CL, Chen M, et al. Antiviral C25 epimers of 26-acetoxy steroids from the South China Sea Gorgonian Echinogorgia rebekka[J]. J Nat Prod, 2014, 77: 1488-1493. DOI:10.1021/np500252q |
| [102] |
Zhang X, Liu Q, Zhang N, et al. Discovery and evolution of aloperine derivatives as novel anti-filovirus agents through targeting entry stage[J]. Eur J Med Chem, 2018, 149: 45-55. DOI:10.1016/j.ejmech.2018.02.061 |
| [103] |
Fois B, Bianco G, Sonar VP, et al. Phenylpropenoids from Bupleurum fruticosum as anti-human rhinovirus species a selective capsid binders[J]. J Nat Prod, 2017, 80: 2799-2806. |
| [104] |
Pirrung MC, Pansare SV, Sarma KD, et al. Combinatorial optimization of isatin-β-thiosemicarbazones as anti-poxvirus agents[J]. J Med Chem, 2005, 48: 3045-3450. DOI:10.1021/jm049147h |