 2020, Vol. 55
2020, Vol. 55



埃博拉病毒(Ebola virus, EBOV)是一种包膜丝状病毒, 其引起的急性出血热是多发于灵长类动物的人畜共患传染病, 其病死率高达50%~90%, 严重时甚至在几天内导致死亡, 是人类危害最严重的传染病之一, 对公共卫生安全和人类的健康有很大的威胁。2014~2016年, 非洲西部暴发了有史以来的一次最严重的埃博拉疫情, 造成上万人死亡, 引起了全球范围的极大恐慌和关注。西非疫情的暴发加速了疫苗的临床研究进展, 重组水泡性口炎疫苗(rVSV-ZEBOV)、黑猩猩腺病毒3型载体疫苗(ChAd3-EBO-Z)以及人腺病毒载体疫苗(Ad26.ZEBOV)相继被发现具有免疫原性[1]。此外多个候选药物也正处于研发阶段。但是目前尚无特效的抗埃博拉出血热治疗药物获准上市, 临床上主要采取对症治疗策略。因此, 研发特异性强、有效性的治疗药物是当务之急, 具有重大的意义。本文在文献调研的基础上, 重点阐述了在埃博拉病毒的复制感染过程中可能的药物靶点, 并根据可能的靶点介绍相应的小分子抑制剂, 然后对几种机制尚不明确的抗埃博拉病毒抑制剂进行了阐述。
1 埃博拉病毒的生物学埃博拉病毒属是单核病毒目丝状病毒科的成员, 包括5种亚型, 即扎伊尔型、苏丹型、科特迪瓦型、本迪布焦型和莱斯顿型[2]。它们在毒力和地理分布方面有显著差异。例如, 莱斯顿埃博拉病毒对人类没有致病性, 而扎伊尔埃博拉病毒是对人类致病性最强的病毒, 致死率高达90%[3]。
埃博拉病毒是一种宽80 nm、长800~1100 nm的线状病毒, 含有1条RNA单链, 该链含有7个基因片段, 每个基因片段都是一个独立的功能单位, 分别编码了7种病毒结构蛋白(图 1)[4]。其中包括基质蛋白VP40和VP24、糖蛋白GP以及RNP复合物NP、VP35、VP30和RNA聚合酶。

|
Figure 1 The structure of the Ebola virus (A) and its genome (B)[4] |
如图 2所示, 埃博拉病毒的感染一般包括吸附、侵入、膜融合、转录翻译、复制、装配、成熟和释放等步骤[5]。首先, 由暴露在病毒表面的GP1, 2介导病毒进入宿主细胞, 完成病毒的吸附和膜融合。随后病毒在宿主细胞内释放遗传物质并进入细胞核, 利用宿主细胞器和原料在病毒RNP复合物的作用下合成子代RNA以及核衣壳。完成病毒的装配后, 装配好的蛋白质和遗传物质在VP40驱动作用下出芽生成病毒颗粒。埃博拉病毒感染的特点是大量产生促炎细胞因子、严重的宿主免疫抑制和快速的病毒血症, 常常表现为暴发性出血。

|
Figure 2 Filamentous virus infection replication cycle[5] |
病毒的复制与增殖传播依赖于病毒与宿主之间的作用, 因此, 从理论上讲, 病毒与宿主之间的相互作用均可作为抗埃博拉病毒小分子药物的靶点。其中, 病毒与宿主之间的相互作用主要包括病毒蛋白与宿主结构蛋白、病毒与宿主酶系统、病毒与宿主信号通路等相互作用。
2.1 干扰病毒感染前期阶段的小分子化合物 2.1.1 靶向糖蛋白GP2018年, Cui等[6]利用建立的重组HIV-1/埃博拉假病毒平台, 评估了一个包含多种喹啉化合物的小分子化合物库, 发现化合物SYL1712 (1)可以有效抑制埃博拉病毒侵入(EC50 = 1 μmol·L-1, CC50 = 214.6 μmol·L-1, SI = 225.9), 并且计算模拟了SYL1712与糖蛋白结合的模型(图 3, 绿色部分为小分子化合物, 蓝色部分为氨基酸残基)。其中, 化合物SYL1712的3-苯基喹啉核心与Tyr517的主链碳原子形成疏水作用, SYL1712末端氮N(CH3)2与Asp522的羧基在3.5Å之内形成强静电作用, 2-苄氧基链与Leu184和Leu186的侧链末端碳原子形成疏水作用。

|
Figure 3 The structure of compound 1 and its binding mode with Ebola glycoprotein[6] |
在晚期内吞体/溶酶体酸性环境下, 病毒的GP1被宿主的组织蛋白酶B和L (CatB/L)切割, 导致GP2构象发生改变, 融合肽插入内吞体膜, 开启内吞体与病毒的膜融合过程[7]。埃博拉病毒膜糖蛋白(GP1, 2)的蛋白水解是膜融合过程中一个重要的步骤。相应地, 病毒表面糖蛋白的水解酶是抑制病毒与宿主融合的重要靶标。非选择性半胱氨酸蛋白酶抑制剂E-64d (2)、选择性组织蛋白酶B (CatB)抑制剂CA-074 (3)、混合组织蛋白酶B/L抑制剂FY-DMK (4)、组织蛋白酶L (CatL)抑制剂Z-FY (t-Bu)-DMK (5)均可以阻碍埃博拉病毒GP1, 2的蛋白水解, 从而减少埃博拉病毒的复制[7, 8]。随后发现, 半胱氨酸蛋白酶抑制剂化合物6和7能够分别减少巨噬细胞[9]和人胚胎肾293T细胞中的埃博拉病毒感染(EC50 = 193 ± 39 nmol·L-1)[10]。此外, 一项基于CatL的埃博拉病毒GP1, 2衍生肽降解的实验发现, 三嗪化合物8和9是CatL抑制剂, 可以减少携带埃博拉病毒GP1, 2的假病毒侵入宿主细胞, 从而发挥抗病毒活性(EC50分别为15和10 μmol·L-1, CC50分别为400和350 μmol·L-1)[11]。
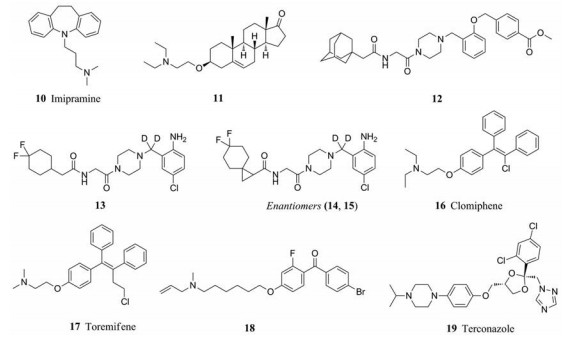
2.1.3 靶向内吞体/溶酶体胆固醇转运蛋白Niemann-Pick C12011年, Carette等[12]利用单倍体遗传筛查技术(haploid genetic screens)证明了内吞体/溶酶体胆固醇转运蛋白Niemann-Pick C1 (NPC1)是埃博拉病毒侵入细胞所必需的关键宿主因子。基因操作对NPC1功能(NPC1表型)的破坏使得体内外均产生对埃博拉病毒的完全抵抗。丙咪嗪(10)和U18666A (11)两种化合物分别通过直接靶向NPC1通路或酸性鞘磷脂酶(ASMase), 在体外降低埃博拉病毒的传染性[13, 14]。与此同时哈佛医学院Cunningham教授实验室通过高通量筛选及结构修饰发现金刚烷二肽哌嗪类化合物12通过阻断EBOV GP1, 2与NPC1的结合, 从而抑制假病毒EBOV-GP1, 2侵入细胞[15], 继而通过亲和标记实验证实了NPC1为化合物12的靶蛋白[16]。它体外抑制埃博拉病毒的活性达到纳摩尔水平, 但较差的药代动力学性质限制了它作为候选药物的开发。
为了改善化合物12的成药性, 2018年, 本课题组与Cunningham教授实验室合作对化合物12进行了多轮结构优化, 通过氘代药物设计策略和手性拆分得到化合物13~15, 在非洲绿猴肾细胞中抑制埃博拉病毒侵入的EC50分别为200、69和27 nmol·L-1, 其体外药代动力学性质相比化合物12有明显的提高(表 1)。除此之外, 小鼠体内药代动力学性质较好, 值得进一步开发[17, 18]。
| Table 1 In vitro pharmacokinetic properties of compounds. a: Kinetic solubility; b: Mouse liver microsome metabolism half life; c: Human liver microsome metabolism half life; d: Mouse plasma protein binding rate; e: Mouse plasma stability (over 4 h) |
2013年, Johansen等[19]运用药物再定位策略, 对美国FDA批准的上市药物进行筛选, 发现选择性雌激素受体(ER)调节剂氯米芬(clomiphene, 16)和托瑞米芬(toremiphene, 17)对埃博拉病毒感染有体外抑制作用, 在Vero E6细胞中, EC50分别为2.42和0.162 μmol·L-1。对感染埃博拉病毒1 h后的C57Bl/6小鼠分别给予氯米芬和托瑞米芬(60 mg·kg-1), 28天存活率分别为90%和50%。氯米芬和托瑞米芬的体外抗病毒作用受到NPC1过度表达的影响。与氯米芬、托瑞米芬类似, 几种阳离子两亲小分子化合物如18、特康唑(19)分别抑制胆固醇合成途径中的2, 3氧化角鲨烯环化酶和羊毛甾醇C14-脱甲基酶, 诱导胆固醇在核内积累, 使NPC1过度表达从而抑制了埃博拉病毒的侵入过程(EC50分别为1.74和7.07 μmol·L-1)。值得注意的是, 与化合物12的结果相反, 所有被测试的阳离子两亲体都没有破坏埃博拉病毒-GP1, 2-NPC1, 这表明该类化合物通过另一种途径干扰NPC1通路[20]。

|
|
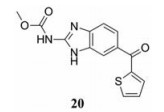
2004年, Yonezawa等[21]发现埃博拉病毒侵入宿主细胞并与宿主细胞融合的过程依赖于功能性细胞骨架, 微管稳定剂紫杉醇可以增强埃博拉病毒的进入, 而诺考达唑(20)可以干扰微管的形成。在37 ℃下用诺考达唑(10 μmol·L-1)或紫杉醇(20 μmol·L-1)处理细胞1 h, 然后用抗α-微管蛋白抗体进行免疫染色, 观察到诺考达唑以剂量依赖性方式抑制埃博拉GP介导的进入和融合。
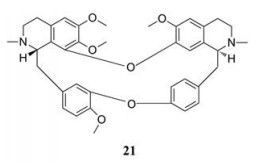
2.1.5 靶向离子通道2015年Sakurai等[22]发现, 埃博拉病毒需经宿主细胞的双孔通道(TPCs)的钙通道才能成功侵入宿主(这些钙通道似乎负责控制含有病毒颗粒的核内体的运动)。因此, 通过阻断或破坏TPCs, 可阻止埃博拉病毒进入宿主细胞, 从而阻止其进一步感染。在阻断TPCs疗法中, 50%的小鼠能够在埃博拉病毒感染中存活下来。TPCs被证明是抗埃博拉病毒的有效靶点, 粉防己碱(21)是该靶点最有效的抑制剂(EC50 = 55 nmol·L-1)[23]。由于粉防己碱也能影响远亲丝状病毒马尔堡病毒的侵入, 推测所有丝状病毒的感染可能都需要TPC的参与。
|
|
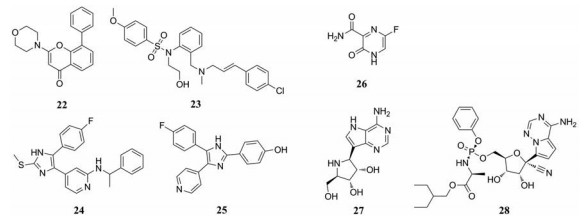
2009年, Kolokoltsov等[24]对激肽组基因产物的siRNA进行了筛选, 结果显示, 3种信号通路蛋白:有丝分裂原活化蛋白激酶(MAPK)、磷酸肌肽3激酶(PI3K)和钙/钙调蛋白激酶(CAMK2)与EBOV病毒颗粒的摄取有关。相应的激酶抑制剂可通过抑制病毒颗粒的摄取, 阻碍GP介导的病毒的进入。PI3K抑制剂LY294002 (22)和CAMK2抑制剂KN-93 (23)在Vero E6细胞水平抑制埃博拉假型病毒的EC50值分别为6.5 ± 1.1和21.4 ± 1.1 μmol·L-1。此外, 一系列吡啶酰咪唑类p38MAPK抑制剂, 如p38inK Ⅲ (24)和SB202190 (25)在PMA分化的巨噬细胞样人THP-1细胞中抑制埃博拉病毒的EC50分别为8.26和4.73 μmol·L-1 [25]。
|
富山化学公司研发的T-705 (favipiravir, 26)具有广谱抗病毒活性, 且细胞毒性较低[26]。T-705通过活性代谢物抑制病毒RNA依赖的RNA聚合酶, 并诱导高致命性RNA突变率[27, 28]。T-705被整合进入宿主细胞后, 在宿主细胞酶的作用下经两次磷酸化转化为三磷酸形式的核糖呋喃酰基磷酸盐(favipiravir-RTP), 抑制病毒RNA聚合酶活性, 从而阻断病毒基因的复制(图 4)。Oestereich等[29]研究发现, 在实验条件下, T-705可抑制扎伊尔型埃博拉病毒在Vero E6细胞中的复制, 其EC50和EC90分别为67和110 μmol·L-1, 无明显细胞毒性。

|
Figure 4 Antiviral mechanism of T-705[30] |
化合物BCX4430 (27)是一种腺苷类似物, 可快速代谢为其磷酸衍生物, 当它与病毒RNA结合时, 该物质可作为RNA链终止物。BCX4430对埃博拉病毒具有体外抑制活性, EC50为11.8 μmol·L-1, CC50大于100 μmol·L-1。与此同时, 在Ames试验中, BCX4430未见致突变性[31]。BCX4430具有与核苷酸相似的药代动力学特征, 在体内可以被迅速清除(在大鼠体内t1/2为5 min), 其磷酸化代谢物清除缓慢(在大鼠体内t1/2为6.2 h)。
由中国军事科学院研制的小分子化合物jk-05 (结构未公布)具有广谱的体内外抗病毒活性(具体活性未公布), 能选择性抑制埃博拉病毒的RNA聚合酶, 从而抑制病毒的复制, 其Ⅰ期临床试验已经完成。2014年, jk-05被中国人民解放军总后勤部批准仅用于军队治疗埃博拉感染的紧急情况[32]。
2016年, Warren等[33]报道了一种新型小分子GS-5734 (28), 它是一种腺苷类似物的单磷酸氨基甲酯前体药物, 通过在细胞内转化为三磷酸核苷(NTP)抑制病毒RNA依赖性RNA聚合酶的活性发挥抗病毒作用。GS-5734在多种人类细胞类型水平(包括原代巨噬细胞和人类内皮细胞)的活性测试中, 均表现出对埃博拉病毒的选择性抑制作用, EC50为0.06至0.14 μmol·L-1, CC50大于1.3 μmol·L-1。此外, 在致死性埃博拉病毒感染的非人类灵长类动物模型中GS-5734取得了显著的抗病毒效果。这是第一个在非人类灵长类动物模型中使用小分子化合物对埃博拉病毒进行实质暴露后保护的案例。目前正在对健康的人体志愿者进行多剂量的GS-5734静脉注射研究, 以评估其临床安全性和药代动力学。
2.2.2 靶向VP352014年, 美国华盛顿大学的研究者首次通过针对VP35IID结构域NP结合位点附近的口袋进行了虚拟筛选, 发现共有20个eIID氨基酸残基组成了这个T型口袋, 其中Gln224、Lys251、Lys248、Ile295、Ile297与吡咯烷酮类化合物GA017 (29)之间产生疏水作用, Gln241的氨基与4位羰基形成氢键(图 5)[34]。体外实验证实此类化合物结合eVP35IID (KD < 100 μmol·L-1), 能够较好地抑制VP35与NP的结合。

|
Figure 5 The binding mode of compound 29[34] |
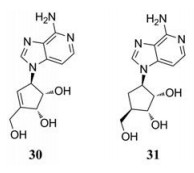
病毒完成转录后需依赖宿主细胞中S-腺苷-L高半胱氨酸水解酶(SAH)对其5′端进行加帽处理, 否则mRNA便无法启动病毒蛋白的翻译。1991年和1995年, Huggins等[35, 36]先后发现两种S-腺苷同型半胱氨酸水解酶抑制剂C-c3Ado (30)和c3Nep (31)具有体外抗埃博拉病毒活性, 在Vero E6细胞水平, EC50分别为30和2 μmol·L-1, CC50分别为5 640和1 700 μmol·L-1。在体内抗病毒活性实验中, 每天两次剂量的C-c3Ado或c3Nep可以延长感染埃博拉病毒的SCID小鼠的生存期。对C-c3Ado的进一步研究证实了这些初步结果:当从第0天或第1天使用C-c3Ado, 由于感染埃博拉病毒的BALB/c小鼠受到剂量依赖的保护, 使得感染小鼠全部存活。从感染到开始治疗之间的时间越长, 存活率和病毒保护越低(C-c3Ado分别在第2天和第3天开始剂量时, 存活率和病毒保护分别为90%和40%)[36]。在使用c3Nep时, 由于开始用药时间的延迟, 疗效也出现了类似的降低[37]。
|
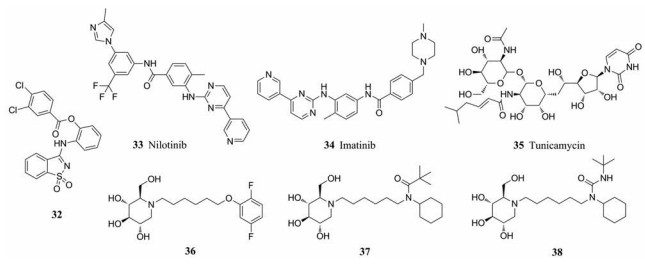
VP40是EBOV的基质蛋白, 在病毒颗粒中含量最多。在VP40的多个结构域中, L-结构域对病毒类似颗粒的高效生产和病毒出芽具有重要作用。在病毒复制后期, 宿主蛋白Nedd4E3、Tsg101与病毒VP40L-结构域相互作用从而保证病毒顺利出芽。因此VP40-Nedd4E3、VP40-Tsg101相互作用是抗EBOV药物设计的重要靶标。以VP40-Tsg101相互作用为靶标, 运用计算机辅助药物设计得到化合物32。在双分子互补法(BiMC)和VLP出芽实验中, 用单独的载体DMSO或浓度递增的化合物32处理人293T细胞1 h, 然后评估和定量VP40 VLP的出芽。研究发现, 化合物32在较低浓度下可抑制约50%的VLP出芽, 在较高浓度下抑制约90%的VLP出芽[38]。
2.3.2 靶向酪氨酸激酶VP40作为主导病毒出芽的核心蛋白, 需要宿主c-Abl1酪氨酸激酶的磷酸化激活后才能发挥其作用。因此, c-Abl1激酶抑制剂尼罗替尼(33)和伊马替尼(34)可减少病毒颗粒的释放, 用伊马替尼或尼罗替尼孵育人293T细胞12至18 h后, 通过NP和VP40蛋白水平检测, 发现VLP释放量呈剂量依赖性降低[39]。
2.3.3 靶向蛋白糖基化埃博拉病毒GPs是病毒成熟的关键要素, 也是病毒出芽的关键媒介, 它们的结构和功能依赖于糖基化。例如, 使用N-链糖基化抑制因子衣霉素(35)治疗, 可将HeLa细胞的EBOV感染降低90%[40]。据报道, 一系列亚胺糖化合物36~38可以通过抑制内质网(ER) α-糖苷酶I (IC50分别为0.09 ± 0.02、0.24 ± 0.08、0.48 ± 0.17 μmol·L-1)而发挥抗埃博拉病毒活性。当埃博拉病毒感染C57Bl/6小鼠4 h后分别给予3种亚胺糖, 小鼠均可达到50%~80%的存活率[41]。
3 机制不明确的小分子化合物以抗埃博拉病毒活性为导向的高通量筛选是快速发现活性先导化合物的主要技术, 但是化合物作用机制的研究往往滞后于化合物的发现, 与此同时, 活性化合物具体作用靶标的确证工作难度较大, 有大量在体内外均具有良好抗病毒活性的化合物的作用靶标尚未明确, 因此目前的研究只能够推测其作用于病毒感染的某个阶段。
FGI系列化合物是通过基于EBOV绿色荧光蛋白报告病毒的高通量筛选得到的一系列具有良好体内外抗EBOV活性的抑制剂。FGI-103 (39)在体外对埃博拉病毒的EC90值为330 nmol·L-1, 细胞毒性小, 小鼠的半数致死剂量大于200 mg·kg-1。研究表明, 该化合物的疗效良好, 并且具有显著的剂量及时间依赖性[42]。FGI-104 (40)是阿莫地喹的类似物, 在Vero E6细胞水平EC50为10 μmol·L-1[43]。FGI-106 (41)是脱氮二苯并萘类抑制剂, 在Vero E6细胞水平EC90为0.9 μmol·L-1。在感染小鼠模型中具有良好的预防及治疗效果, 5 mg·kg-1的预防给药剂量即能够达到100%的保护率。目前推测FGI-106的抗病毒机制可能是作用于宿主的信号通路, 继而影响病毒RNA的合成[44]。FGI-106的结构衍生物42在体外同样具有良好的抗EBOV活性, 其EC50值为1.13 ± 0.28 μmol·L-1, 且无明显的细胞毒性[45]。
|
2013年Madrid等[46]基于药物重定位筛选策略, 发现氯喹(43)对病毒的侵入与复制均产生影响, 在埃博拉假病毒侵入实验中, 氯喹在HEK 293T细胞水平的EC50和EC90分别为4.7和43 μmol·L-1。病毒复制实验中, 氯喹在Vero 76细胞水平的EC50和EC90分别为16和35 μmol·L-1。此外雄性或雌性C57BL/6和Balb/c小鼠体内研究显示, 在感染前4 h以90 mg·kg-1剂量给药时, 氯喹能显著降低死亡率(在第13天存活率为90%)。
除了上述埃博拉病毒抑制剂, 还有几种不同类型的化合物可以影响埃博拉病毒的复制, 包括热休克蛋白90 (HSP90)抑制剂17-AAG[47]、钠/钾转运ATP酶亚单位alpha-1 (ATP1A1)抑制剂乌本苷和11-羟基甾体脱氢酶抑制剂甘草酸。此外, 异噁唑衍生物44可抑制EBOV GP1, 2-伪型HIV颗粒侵入293T细胞(EC50 = 2.5 μmol·L-1)[48]。
4 总结与展望综上, 迄今为止仍然没有防治埃博拉病毒感染的特异性药物。通过对近年来抗埃博拉病毒小分子抑制剂的回顾发现, 为了改变这一现状, 研究人员致力于发现新机制与新结构的抗埃博拉先导化合物, 在小分子化合物库及天然产物库的构建及活性筛选方面付出了大量努力, 取得了一系列进展。值得关注的是, 对现有药物再定位(即老药新用)是快速找到抗病毒特效药物的有效方法, 这些“老药”可以直接跳过临床Ⅰ期, 从而大大降低研发成本, 并且后期不良反应风险性也比较低。此外, 计算机辅助药物设计是发现抗埃博拉病毒活性先导化合物的重要手段, 即根据埃博拉病毒蛋白的功能特征和三维结构, 确定配体结合位点和结合模式, 基于受体进行筛选找到苗头化合物。在人工智能时代, 机器学习算法必将加速抗埃博拉病毒新药的研发。

|
需要指出的是, 抗埃博拉病毒活性测试实验需要在生物安全4级实验室内进行, 国内具备此级别的实验室并不多, 因此制约了有关埃博拉病毒的基础研究。但随着反向遗传和真核细胞蛋白表达技术的发展, 通过构建复制型埃博拉病毒颗粒进行病毒活性测试弥补了上述实验条件的缺陷, 促进了埃博拉病毒研究的进一步发展。随着病毒蛋白功能研究的不断完善, 病毒感染复制过程中的宿主互作因子逐渐引起人们的关注。针对宿主互作因子开发新型抗病毒药物具有明显优势, 可以大大克服病毒的耐药性。因此未来研究的重点除了对现有抗埃博拉病毒先导化合物的结构优化, 还应基于埃博拉病毒宿主互作因子结构与生物学的认识, 开发出结构新颖、活性突出的抗埃博拉病毒药物。
| [1] |
Malvy D, McElroy AK, de Clerck H, et al. Ebola virus disease[J]. Lancet, 2019, 393: 936-948. DOI:10.1016/S0140-6736(18)33132-5 |
| [2] |
Li YH, Chen SP. Evolutionary history of Ebola virus[J]. Epidemiol Infect, 2014, 142: 1138-1145. DOI:10.1017/S0950268813002215 |
| [3] |
Feldmann H, Geisbert TW. Ebola haemorrhagic fever[J]. Lancet, 2011, 377: 849-862. DOI:10.1016/S0140-6736(10)60667-8 |
| [4] |
Picazo E, Giordanetto F. Small molecule inhibitors of ebola virus infection[J]. Drug Discov Today, 2015, 20: 277-286. DOI:10.1016/j.drudis.2014.12.010 |
| [5] |
Messaoudi I, Amarasinghe GK, Basler CF. Filovirus pathogenesis and immune evasion: insights from Ebola virus and Marburg virus[J]. Nat Rev Microbiol, 2015, 13: 663-676. DOI:10.1038/nrmicro3524 |
| [6] |
Cui Q, Cheng H, Xiong R, et al. Identification of diaryl-quinoline compounds as entry inhibitors of Ebola virus[J]. Viruses, 2018, 10: 678. DOI:10.3390/v10120678 |
| [7] |
Chandran K, Sullivan NJ, Felbor U, et al. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection[J]. Science, 2005, 308: 1643-1645. DOI:10.1126/science.1110656 |
| [8] |
Schornberg K, Matsuyama S, Kabsch K, et al. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein[J]. J Virol, 2006, 80: 4174-4178. DOI:10.1128/JVI.80.8.4174-4178.2006 |
| [9] |
Gnirss K, Kuhl A, Karsten C, et al. Cathepsins B and L activate Ebola but not Marburg virus glycoproteins for efficient entry into cell lines and macrophages independent of TMPRSS2 expression[J]. Virology, 2012, 424: 3-10. DOI:10.1016/j.virol.2011.11.031 |
| [10] |
Shah PP, Wang T, Kaletsky RL, et al. A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and ebola pseudotype virus infection into human embryonic kidney 293T cells[J]. Mol Pharmacol, 2010, 78: 319-324. |
| [11] |
Elshabrawy HA, Fan J, Haddad CS, et al. Identification of a broad-spectrum antiviral small molecule against severe acute respiratory syndrome coronavirus and Ebola, Hendra, and Nipah viruses by using a novel high-throughput screening assay[J]. J Virol, 2014, 88: 4353-4365. DOI:10.1128/JVI.03050-13 |
| [12] |
Carette JE, Raaben M, Wong AC, et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1[J]. Nature, 2011, 477: 340-343. DOI:10.1038/nature10348 |
| [13] |
Cenedella RJ. Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes[J]. Lipids, 2009, 44: 477-487. DOI:10.1007/s11745-009-3305-7 |
| [14] |
Rodriguez-Lafrasse C, Rousson R, Bonnet J, et al. Abnormal cholesterol metabolism in imipramine-treated fibroblast cultures. Similarities with Niemann-Pick type C disease[J]. Biochim Biophys Acta, 1990, 1043: 123-128. DOI:10.1016/0005-2760(90)90284-5 |
| [15] |
Cote M, Misasi J, Ren T, et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection[J]. Nature, 2011, 477: 344-348. DOI:10.1038/nature10380 |
| [16] |
Lee K, Ren T, Cote M, et al. Inhibition of Ebola virus infection: identification of Niemann-Pick C1 as the target by optimization of a chemical probe[J]. ACS Med Chem Lett, 2013, 4: 239-243. DOI:10.1021/ml300370k |
| [17] |
Tian Y. Study on N-Containing Heterocyclic HIV-1 NNRTI and Ebola Virus Entry Inhibitors (含N芳杂环类HIV-1非核苷类逆转录酶抑制剂及埃博拉病毒侵入抑制剂的研究) [D]. Jinan: Shandong University, 2017.
|
| [18] |
Liu H, Tian Y, Lee K, et al. Identification of potent Ebola virus entry inhibitors with suitable properties for in vivo studies[J]. J Med Chem, 2018, 61: 6293-6307. DOI:10.1021/acs.jmedchem.8b00704 |
| [19] |
Johansen LM, Brannan JM, Delos SE, et al. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection[J]. Sci Transl Med, 2013, 5: 190ra179. |
| [20] |
Shoemaker CJ, Schornberg KL, Delos SE, et al. Multiple cationic amphiphiles induce a Niemann-Pick C phenotype and inhibit Ebola virus entry and infection[J]. PLoS One, 2013, 8: e56265. DOI:10.1371/journal.pone.0056265 |
| [21] |
Yonezawa A, Cavrois M, Greene WC. Studies of Ebola virus glycoprotein-mediated entry and fusion by using pseudotyped human immunodeficiency virus type 1 virions: involvement of cytoskeletal proteins and enhancement by tumor necrosis factor alpha[J]. J Virol, 2005, 79: 918-926. DOI:10.1128/JVI.79.2.918-926.2005 |
| [22] |
Sakurai Y, Kolokoltsov AA, Chen CC, et al. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment[J]. Science, 2015, 347: 995-998. |
| [23] |
Zhu MX, Ma J, Parrington J, et al. Calcium signaling via two-pore channels: local or global, that is the question[J]. Am J Physiol Cell Physiol, 2010, 298: C430-441. DOI:10.1152/ajpcell.00475.2009 |
| [24] |
Kolokoltsov AA, Saeed MF, Freiberg AN, et al. Identification of novel cellular targets for therapeutic intervention against Ebola virus infection by siRNA screening[J]. Drug Dev Res, 2009, 70: 255-265. |
| [25] |
Johnson JC, Martinez O, Honko AN, et al. Pyridinyl imidazole inhibitors of p38 MAP kinase impair viral entry and reduce cytokine induction by Zaire ebolavirus in human dendritic cells[J]. Antiviral Res, 2014, 107: 102-109. DOI:10.1016/j.antiviral.2014.04.014 |
| [26] |
Furuta Y, Takahashi K, Fukuda Y, et al. In vitro and in vivo activities of anti-influenza virus compound T-705[J]. Antimicrob Agents Chemother, 2002, 46: 977-981. DOI:10.1128/AAC.46.4.977-981.2002 |
| [27] |
Sangawa H, Komeno T, Nishikawa H, et al. Mechanism of action of T-705 ribosyl triphosphate against influenza virus RNA polymerase[J]. Antimicrob Agents Chemother, 2013, 57: 5202-5208. DOI:10.1128/AAC.00649-13 |
| [28] |
Espy N, Nagle E, Pfeffer B, et al. T-705 induces lethal mutagenesis in Ebola and Marburg populations in macaques[J]. Antiviral Res, 2019, 170: 104529. |
| [29] |
Oestereich L, Ludtke A, Wurr S, et al. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model[J]. Antiviral Res, 2014, 105: 17-21. DOI:10.1016/j.antiviral.2014.02.014 |
| [30] |
Furuta Y, Komeno T, Nakamura T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase[J]. Proc Jpn Acad Ser B Phys Biol Sci, 2017, 93: 449-463. DOI:10.2183/pjab.93.027 |
| [31] |
Warren TK, Wells J, Panchal RG, et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430[J]. Nature, 2014, 508: 402-405. DOI:10.1038/nature13027 |
| [32] |
Gao J, Yin L. Drug development for controlling Ebola epidemic - a race against time[J]. Drug Discov Ther, 2014, 8: 229-231. DOI:10.5582/ddt.2014.01040 |
| [33] |
Warren TK, Jordan R, Lo MK, et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys[J]. Nature, 2016, 531: 381-385. DOI:10.1038/nature17180 |
| [34] |
Brown CS, Lee MS, Leung DW, et al. In silico derived small molecules bind the filovirus VP35 protein and inhibit its polymerase cofactor activity[J]. J Mol Biol, 2014, 426: 2045-2058. DOI:10.1016/j.jmb.2014.01.010 |
| [35] |
de Clercq E, Montgomery JA. Broad-spectrum antiviral activity of the carbocyclic analog of 3-deazaadenosine[J]. Antiviral Res, 1983, 3: 17-24. DOI:10.1016/0166-3542(83)90011-6 |
| [36] |
Huggins J, Zhang ZX, Bray M. Antiviral drug therapy of filovirus infections: S-adenosylhomocysteine hydrolase inhibitors inhibit Ebola virus in vitro and in a lethal mouse model[J]. J Infect Dis, 1999, 179: S240-247. |
| [37] |
Bray M, Driscoll J, Huggins JW. Treatment of lethal Ebola virus infection in mice with a single dose of an S-adenosyl-L-homocysteine hydrolase inhibitor[J]. Antiviral Res, 2000, 45: 135-147. DOI:10.1016/S0166-3542(00)00066-8 |
| [38] |
Liu Y, Lee MS, Olson MA, et al. Bimolecular complementation to visualize filovirus VP40-host complexes in live mammalian cells: toward the identification of budding inhibitors[J]. Adv Virol, 2011, 2011. |
| [39] |
Garcia M, Cooper A, Shi W, et al. Productive replication of Ebola virus is regulated by the c-Abl1 tyrosine kinase[J]. Sci Transl Med, 2012, 4: 123ra124. |
| [40] |
Chan SY, Speck RF, Ma MC, et al. Distinct mechanisms of entry by envelope glycoproteins of Marburg and Ebola (Zaire) viruses[J]. J Virol, 2000, 74: 4933-4937. DOI:10.1128/JVI.74.10.4933-4937.2000 |
| [41] |
Chang J, Warren TK, Zhao X, et al. Small molecule inhibitors of ER alpha-glucosidases are active against multiple hemorrhagic fever viruses[J]. Antiviral Res, 2013, 98: 432-440. DOI:10.1016/j.antiviral.2013.03.023 |
| [42] |
Warren TK, Warfield KL, Wells J, et al. Antiviral activity of a small-molecule inhibitor of filovirus infection[J]. Antimicrob Agents Chemother, 2010, 54: 2152-2159. DOI:10.1128/AAC.01315-09 |
| [43] |
Kinch MS, Yunus AS, Lear C, et al. FGI-104: a broad-spectrum small molecule inhibitor of viral infection[J]. Am J Transl Res, 2009, 1: 87-98. |
| [44] |
Aman MJ, Kinch MS, Warfield K, et al. Development of a broad-spectrum antiviral with activity against Ebola virus[J]. Antiviral Res, 2009, 83: 245-251. DOI:10.1016/j.antiviral.2009.06.001 |
| [45] |
Selakovic Z, Opsenica D, Eaton B, et al. A limited structural modification results in a significantly more efficacious diazachrysene-based filovirus inhibitor[J]. Viruses, 2012, 4: 1279-1288. DOI:10.3390/v4081279 |
| [46] |
Madrid PB, Chopra S, Manger ID, et al. A systematic screen of FDA-approved drugs for inhibitors of biological threat agents[J]. PLoS One, 2013, 8: e60579. DOI:10.1371/journal.pone.0060579 |
| [47] |
Smith DR, McCarthy S, Chrovian A, et al. Inhibition of heat-shock protein 90 reduces Ebola virus replication[J]. Antiviral Res, 2010, 87: 187-194. |
| [48] |
Yermolina MV, Wang J, Caffrey M, et al. Discovery, synthesis, and biological evaluation of a novel group of selective inhibitors of filoviral entry[J]. J Med Chem, 2011, 54: 765-781. DOI:10.1021/jm1008715 |